
小花草玉梅高通量转录组测序与花发育基因的挖掘
2018-10-16 05:51戎利勤李晓冬刘虎岐
西北植物学报 2018年8期
戎利勤,李晓冬,刘虎岐
(西北农林科技大学 生命科学学院,陕西杨陵 712100)
花是被子植物独有的观赏及生殖器官,花发育的研究对植株发育及演化过程有重要影响[1]。在拟南芥、金鱼草等模式植物中已克隆到许多与花分生组织形成和花器官形成等相关的基因[2]。Coen等开创性地提出了花器官发育的ABC模型[3]。但此模型是以核心真双子叶植物为基础的。毛茛科属于基部真双子叶植物[4],花器官发育并不严格遵守基于核心真双子叶植物的ABC模型,该类群MADS-box基因调控范围更广,延展到相邻花器官,使花器官形态具有高度多样性[5-6]。
小花草玉梅(Anemonerivularisvar.flore-minore)是毛茛科银莲花属草玉梅的变种[7]。小花草玉梅的花器官在野生条件下发生了变异,变异在花被片、雄蕊和雌蕊中均有体现[8]。根据其花器官变异部位及程度将变异分为5类:全白、绿白相间、五瓣、全绿和极端变异[9]。
Illumina HiSeq 2000第二代高通量测序平台使用PE90技术,在测序速度以及通量方面都得到了优化[10]。本研究通过高通量测序技术Illumina HiSeq 2000,在小花草玉梅中进一步挖掘参与其花形态建成的重要基因。小花草玉梅作为基部真双子叶植物,转录组数据对其变异机制的研究奠定了基础,为人们更全面研究被子植物的系统发育与遗传演化提供基础数据。
1 材料和方法
1.1 试验材料
2016年7月于陕西省陇县地区采集小花草玉梅植株的正常花器官和变异花器官,经液氮速冻后于-80 ℃贮存备用。对正常花器官进行转录组测序分析。
1.2 方 法
1.2.1RNA提取与测序文库的构建按照Trizol法提取样品的总RNA,用带有Oligo(dT)的磁珠富集mRNA。将mRNA打断成片段后逆转录成dsDNA,在经过纯化洗脱之后做末端修复、加polyA并连接测序接头,制备测序文库,富集测序样本。
1.2.2转录组测序通过Illumina HiSeq 2000测序平台进行高通量测序。为得到较高质量的结果,原始数据中要去除带接头的,重复的以及测序质量很低的读序,再用Trinity软件进行转录组重头组装,先将具有一定长度重叠的reads连成更长的不含N的片段Contig,之后将来自同一转录本的不同Contig连接,得到两端不能再延长的非冗余序列。
1.2.3转录组数据分析使用BLAST程序,将小花草玉梅unigenes与NCBI的非冗余蛋白数据库(Non-redundant protein database,NR),去冗余的蛋白序列数据库(SwissProt protein database,SwissProt),蛋白质直系同源数据库(Cluster of Orthologous Groups,COG)和基因功能和代谢途径数据库(Kyoto encyclopedia of genes and genomes,KEGG)等数据库进行Blastx比对,得到与给定unigene具有最高序列相似性的蛋白,得到该unigene的注释信息。根据NR注释信息,使用Blast2GO软件得到基因的基因本体论数据库(Gene Ontology,GO)注释信息。
1.2.4花发育基因的实时定量PCR试验将转录组得到的序列与北京大学的转录因子数据库(Plant TFDB)进行比对分析,筛选出了12个与花发育相关的MADS基因。对12个基因在小花草玉梅正常花和5种变异花(图1,B~F)中做定量反应。5种变异花分别为全白变异(图1,B),绿白相间变异(图 1,C),五瓣变异(图 1,D),全绿变异(图 1,E)和极端变异(图 1,F)。利用Primer5设计定量PCR引物(表1),选择β-actin作为内参基因,使用相对定量2-ΔΔCt法分析。运用SPSS软件对基因表达量进行主成分分析,筛选出参与花形态建成的主要基因。

图1 小花草玉梅正常花(A)和变异花(B~F)的形态Fig.1 Morphology of normal (A) and variant flowers (B~F) in Anemone rivularis var. flore-minore

基因Gene正向引物Forward primer(5'→3')反向引物Reverse primer(5'→3')产物长度The length of the product/bpFUL1AAGCATCCAGGGTGGCATGAGCTTCGTCCCGCTGCAGG121FUL2TGATTAAGCATCCAGGGAGGAATCAGAGGTCCAACTGTGAGGAACATGTTCAAATGG180AP3-1CGATCCGCCAGCACCATCGCCATTAACTACTCACACTGAAACAACCAG169AP3-2CCCGACATGCCACTATGGATTCAAGCAAGGGTTAAACCATATGAGCT151AP3-3CATGTTCTCTTGCACCGGCAATCCATCCCAGTCCTTCGCC199PI1ATCGCATAAACAGGAACAGCAGATGGCAAATTTGGCTGGATTGGCTGGACT136PI2GCACCTCCGAAGGATAGTCTGTACTGGACAATGAACCAGCAGGAAATGAA97AG1CCAAATCTAGCAGCAACGGGAGATACGGCCACGGCTAGAG201AG2GACTTGTTCTGACCCACAAACTGCTGGTTTGAGTTCTGCAGAATTTCAATCTGCTG113SEP1CCTTGGAACATTCATAGGGTATGCATCTGCTCCAAGCACCAAAGAACAGAAAAGATGT205SEP3ACATTTGGCTCTGGTCCTCCTCGCCAAACGTAGAAATGGC184AGL6ATCTCCGCCGCAAGGAGCGTCCGATTTGCAAGGTGGGCTC189β-actinGAGCCCAGAGGTGCTCTTAGCAATGCCAGGGAACATGG165
2 结果与分析
2.1 小花草玉梅转录组数据的组装
对小花草玉梅花器官转录组进行测序,共得到54 513 822个读序(reads),Q20的百分率为95.49%、Q30的百分率为88.90%、GC含量百分比为46.33%。以上结果表明,转录组测序数据质量可靠,可用于后续分析。序列组装后,最终得到了43 767条unigenes,总长度为40 565 399 bp,平均长度与N50分别为926 和1 431 bp。其中,大于2 000 bp的序列共有4 280条,占unigenes总数的9.78%,说明测序质量较好。
2.2 小花草玉梅unigene的功能注释、分类和代谢途径分析
使用BLAST程序将测序得到的unigenes分别与NR、Swissprot、COG、KEGG数据库比对,结果(表2)显示,在NR注释成功的unigenes总unigenes数的百分率最高(64.09%),在KEGG注释成功的unigenes占比则最低,为26.44%。
对4个数据库的注释信息分析,共有9 379条unigenes在所有数据库中同时标注成功,占unigenes总数的21.43%。在所有数据中,只在某一个数据库中注释成功的unigenes为5 105条,比例为11.66%。在以上4个数据库中至少1个数据库注释成功的unigenes有28 130条。用相似序列匹配得到的近缘物种中,莲花(Nelumbonucifera)所占比例最高(28.34%),其次是葡萄(Vitisvinifera,8.53%),可可(Theobromacacao,5.41%)。

表2 小花草玉梅unigenes的功能注释结果
2.3 小花草玉梅的SSR分析
利用MISA软件在小花草玉梅花器官的unigenes中检测到5 015个SSR位点,占unigenes总序列的11.46%。SSR的类型丰富,其中,三核苷酸重复所占比例最高,达到了49.77%;五核苷酸重复所占比例最低,为3.05%;二核苷酸重复、四核苷酸重复和六核苷酸重复所占比例分别为27.32%、6.66%和13.20%。在搜索到的SSR中,出现频率最高的5类基序为:AG/CT(20.7%)、AAG/CTT(15.8%)、ACC/GGT(8.4%)、ATC/ATG(7%)、AGC/CTG(5.1%)。上述SSR特征的分析,有助于开展小花草玉梅花器官的通用性标记开发等研究。
2.4 小花草玉梅花发育基因的定量实验
采用qPCR方法,以小花草玉梅正常花为对照,设定基因的表达量为1,得出各基因的相对表达量,分别对12个MIKC 型 MADS-box 基因在小花草玉梅正常花和变异花中的表达量水平进行了研究,结果(图2)表明,与小花草玉梅正常花相比,全白、绿白相间、五瓣变异、全绿变异花中的FUL1、SEP1,SEP3和AGL6基因均显著上调表达,而12个基因在极端变异花中的表达水平与正常花的差异均不明显。
对12个花发育基因在正常花和5种变异花中的表达量进行主成分分析。按特征值>1的原则,提取了3个主成分,贡献率分别为41.54%、33.62%、13.45%,累计方差贡献率达88.61%,涵盖了大部分信息。由主成分的初始因子载荷矩阵(表3)可以看出,AGL6、SEP3、FUL1、PI2及SEP1的表达量与第1主成分呈显著正相关,AG2、AP3-1、AG1、AP3-3、PI1的表达量与第2主成分呈显著正相关。主成分分析表明,与第1主成分呈显著正相关的指标,即AGL6、SEP3、FUL1、PI2及SEP1的表达量均可作为小花草玉梅花形态发育的主要指标。
图3是小花草玉梅的12个花发育MADS基因的表达量经主成分分析后的前3个主成分构建的三维空间。分析的样本有6个,分别为小花草玉梅正常花和5种变异花。具有不同花形态的小花草玉梅分布在三维空间中不同的位置。6个样品表型不同,各自起主要调控作用的基因也不相同,因此它们位于空间中的不同位置。
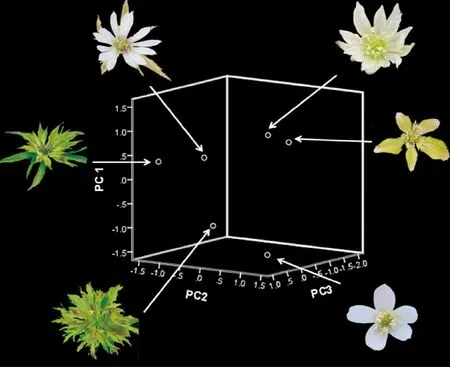
图3 不同形态的小花草玉梅在MADS基因三维空间中的分布位置Fig.3 A. rivularis var. flore-minore species placed in 3D space of MADS gene

图2 12个MIKC 型 MADS-box 基因在小花草玉梅正常花和变异花中的相对表达量Fig.2 Relative expression level of 12 MIKC-type MADS genes in normal and variant flowers in A. rivularis var. flore-minore
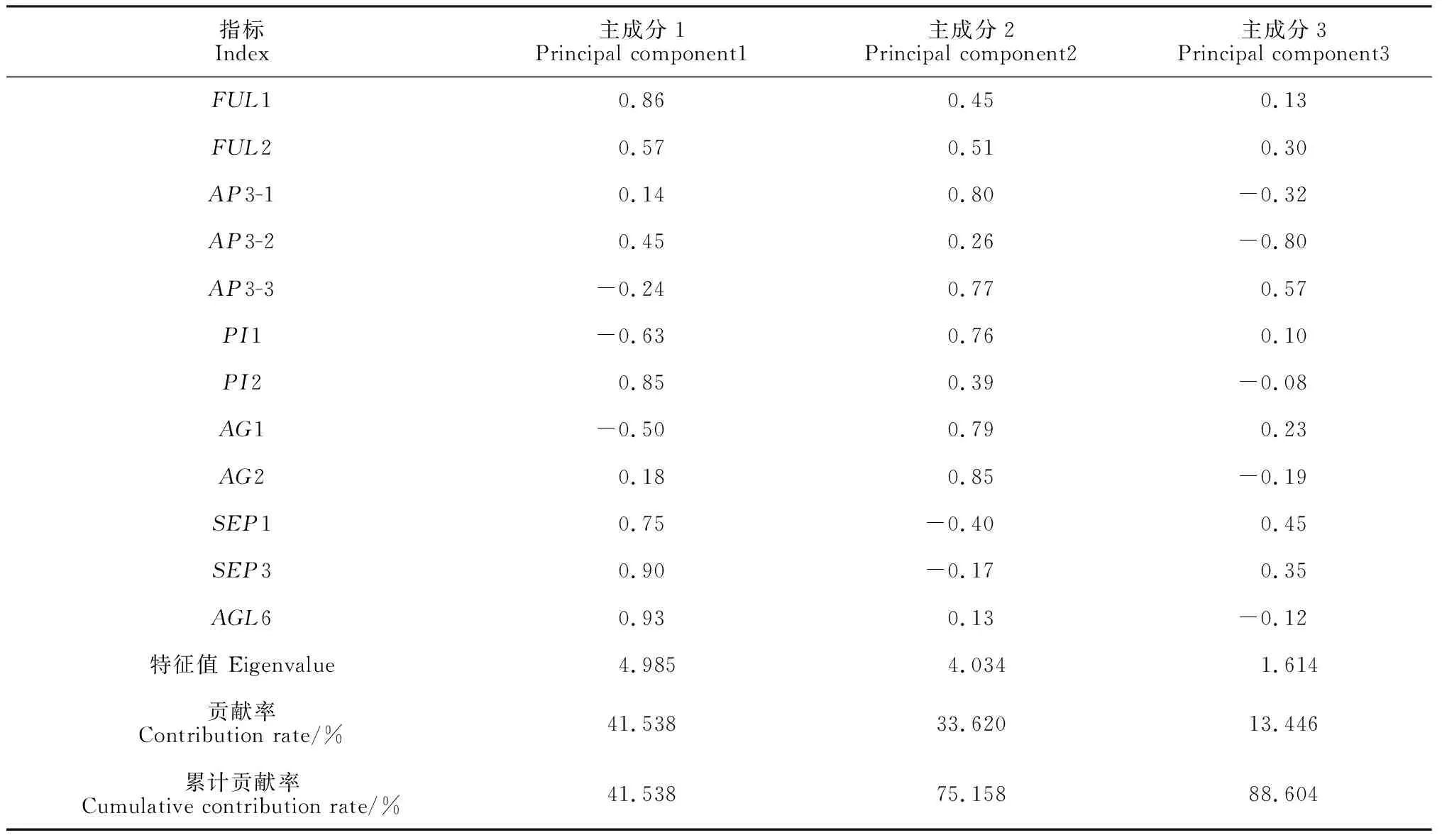
指标Index主成分1Principal component1主成分2Principal component2主成分3Principal component3FUL10.860.450.13FUL20.570.510.30AP3-10.140.80-0.32AP3-20.450.26-0.80AP3-3-0.240.770.57PI1-0.630.760.10PI20.850.39-0.08AG1-0.500.790.23AG20.180.85-0.19SEP10.75-0.400.45SEP30.90-0.170.35AGL60.930.13-0.12特征值 Eigenvalue4.9854.0341.614贡献率 Contribution rate/%41.53833.62013.446累计贡献率Cumulative contribution rate/%41.53875.15888.604
3 讨 论
Illumina高通量测序技术的测序数据量大、效率高且成本低[11]。本研究共得到43 767条unigenes,平均长度为926 bp,高于薏苡幼苗叶片737.85 bp[12]、半夏珠芽751 bp[13],梁山慈竹857.89 bp[14],说明小花草玉梅序列组装效果较好。N50值为1 431 bp,N50值越大说明组装得到的长片段就越多,组装效果就越好[15];Q20的百分率为95.49%、Q30的百分率为88.90%、高于它们的规定限值(>80%),以上研究结果表明,本研究采用的双端测序的方法,增加了测序深度,且提高了拼接的效率和准确性[16]。此次测序质量可靠,可以满足转录组分析的基本要求。
将测序结果分别与NR、Swissprot、KOG、KEGG数据库比对,得到每个数据库注释的unigenes占总unigenes分别为64.09%、48.21%、39.85%和26.44%。在以上4个数据库中至少在1个数据库注释成功的unigenes占比为64.27%。有35.73%的unigenes未得到注释,一方面可能是因为unigenes序列片段长度过短,或是非编码序列,难以进行同源性比对;另一方面,小花草玉梅基因组和转录组遗传信息匮乏,某些未被注释的基因可能是其特有的基因。将数据在NR数据库中进行分析,发现大多数unigenes(28.34%)能够成功匹配到莲花的蛋白质序列,其次是葡萄(8.53%)和可可(5.41%),这可能与目前NCBI数据库中莲花、葡萄和可可的基因组数据比较丰富有关。
在黑种草中,有研究者通过基因敲除技术来研究花器官形成机制,研究表明决定花器官特征的基因有AP3-1、AP3-2、AP3-3、PI1、PI2、AG1、SEP1、SEP2、SEP3和AGL6,它们表达量的不同会影响花形态的建成[17]。本研究中的小花草玉梅花器官在野生条件下发生了变异,王超等的研究将变异分为5类,分别为全白变异、绿白相间变异、五瓣变异、全绿变异和极端变异。张婷等研究发现,正常植株和绿白相间变异植株的AP3-3基因序列是不同的,在变异植株的上游调控区有一段49 bp的插入;突变植株的AP3-3序列与正常相比有4个碱基突变点[18]。参照前人对小花草玉梅的研究,本研究选取了12个花发育相关的MADS基因分别在小花草玉梅正常花和5种变异花中做实时定量PCR实验,通过主成分分析表明,AGL6、SEP3、FUL1、PI2及SEP1的表达量为小花草玉梅花形态建成的主要影响指标。
本研究首次建立了小花草玉梅转录组数据库,有助于其分子遗传信息的大量扩充,初步挖掘了12条花发育相关的MADS基因,为进一步研究小花草玉梅花形态发育及变异的分子机制,开展花发育的基因克隆及功能验证等研究提供了宝贵资源。
猜你喜欢
流行色(2021年8期)2021-11-09
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
汉语世界(The World of Chinese)(2019年5期)2019-11-11
小天使·一年级语数英综合(2019年4期)2019-10-06
小学生优秀作文(低年级)(2018年3期)2018-08-15
少儿科学周刊·少年版(2018年12期)2018-01-26
科学中国人(2017年36期)2017-06-09
Coco薇(2017年5期)2017-06-05
琴童(2016年7期)2016-05-14
