
药物中磺酸酯类基因毒性杂质研究进展
2018-10-11 05:25刘雪薇韩海云张文鹏陈东英
色谱 2018年10期
刘雪薇, 厉 程, 韩海云, 张文鹏, 陈东英*
(1. 中国科学院上海药物研究所, 上海 201203; 2. 中国科学院大学, 北京 100049; 3. 上海药品审评核查中心, 上海 201203)
基因毒性杂质是指在药物中以痕量水平存在,能够和DNA发生反应诱导DNA损伤,导致基因突变并有可能诱发癌症的杂质[1,2]。与药物中其他类型的杂质相比,基因毒性杂质具有在极低暴露水平下即能导致严重毒性的特点,因此对用药的安全性造成严重的威胁。磺酸酯是一类潜在的基因毒性杂质,通常来源于药物合成中磺酸或者其衍生物,如磺酰氯、磺酸酐和低级醇溶剂(如甲醇、乙醇、异丙醇等)之间发生的副反应。由于磺酸盐药物应用非常广泛且其在合成中不可避免地会用到低级醇,使得磺酸酯在药物中的残留风险较为常见,因而备受人们关注。本文将首先对基因毒性杂质相关的监管法规进行梳理,进一步对磺酸酯类基因毒性杂质的形成机理,控制策略以及相关的分析方法进行综述。
1 基因毒性杂质监管理念的演变
针对基因毒性杂质给用药带来的安全隐患,各国监管机构的监管理念主要经历了由追求完全避免、合理降低再到风险评估3个主要发展历程[3,4]。
欧洲药品管理局(European Medicines Agency, EMA)[5]最先出台了关于基因毒性杂质的控制法规,其下属的人用药物委员会(原专利药物委员会)于2002年发布了一份关于基因毒性杂质限度要求的咨询意见书(Position paper on the limits of genotoxic impurities)。该意见书保守假设了基因毒性杂质在任何暴露水平均能造成DNA损伤,因此任何浓度水平的基因毒性杂质都会给用药安全带来风险。基于此假设,该意见书要求制药企业尽可能寻求不会产生基因毒性杂质的“零风险”工艺路线;在达不到“零风险”要求的情况下,制药企业需要提供足够充分的理由,解释无法避免基因毒性杂质的原因,并在技术水平能达到的情况下尽可能地减少(as low as technically feasible, ALATF)药物中基因毒性杂质的残留量。这份意见书给基因毒性杂质的控制提供了初步的实施方案和标准,但是它的保守性甚至是极端性也给制药企业带来了很大的挑战,在一定程度上阻碍了涉及相关药物的研发进程。在该意见书的基础上,EMA和制药企业进行了多轮探讨,于2006年颁布了《基因毒性杂质限度指南》(Guideline on the limits of genotoxic impurities)[6]。该指南不再追求难以实现的“零风险”,首次引入了毒理学关注阈值(threshold of toxicological concern, TTC)的概念,并采用“最低合理可行”(as low as reasonably practical, ALARP)原则代替ALATF。TTC是指由引起50%肿瘤发生率(TD50值,由最敏感物种的最敏感肿瘤部位获得),通过简单线性外推至十万分之一肿瘤发生率所对应的基因毒性杂质暴露量(不考虑DNA修复、细胞保护等因素带来的影响)。通过线性外推,TTC值设定为1.5 μg/d,意味着当每人每天所摄入的基因毒性杂质的量小于1.5 μg时,人一生中(按寿命为70年计算)患癌症的风险将小于十万分之一。与之前寻求“零风险”的做法相比,TTC概念的引入对基因毒性杂质的控制来说有了长足进步,标志着对药物中基因毒性杂质的控制进入了风险评估的发展阶段。然而,无论长期用药还是短期用药,该指南都以1.5 μg/d 作为基因毒性杂质的控制限度,该限度水平对于终生服药的患者来说是可以接受的,但是对于短期暴露的药物来说显然还是过于保守。2006年1月,由辉瑞、强生、葛兰素史克等国际制药企业中的研发人员组成的美国药物研究和制造商协会(Pharmaceutical Research and Manufacturers of America, PhRMA)专家小组在Lutz Müeller等的领导下首次提出了“阶段化TTC”的概念,即原料药中的基因毒性杂质限度应当考虑药物的暴露周期,暴露周期较短的药物应当可以设定较高的控制限度[7]。“阶段化TTC”概念是风险评估科学理念的进一步体现,它的提出使得人们能够根据用药周期,科学、灵活地开展基因毒性杂质的控制。
基因毒性杂质通常含有某些特定的化学结构单元而具有高反应活性,在体内转化为高的生物活性,从而与DNA等功能分子发生反应而引起毒性。这些特定的化学结构单元具有基因毒性的警示性,被称为警示结构[8,9]。常见的警示结构主要包括芳香胺、醛、N-亚硝基胺、氨基甲酸类、环氧丙烷、氮杂环丙烷、肼、卤代烷烃和磺酸酯等类型(见图1)。Müeller等[7]将药物中的杂质按照结构特性及诱变性和致癌性分成了5类,并提出了相应的控制策略(见表1),为新药研发及具体监管的实施给出了更为合理的权衡方案。

图 1 基因毒性杂质警示结构[7]Fig. 1 Alerting structures of genotoxic impurities[7] A: alkyl, aryl, or H; R: alkyl; EWG: electron withdrawing group (CN, C=O, ester, etc); PNAs: p-nitroaniline; PNAHs: polycyclic aromatic hydrocarbon.
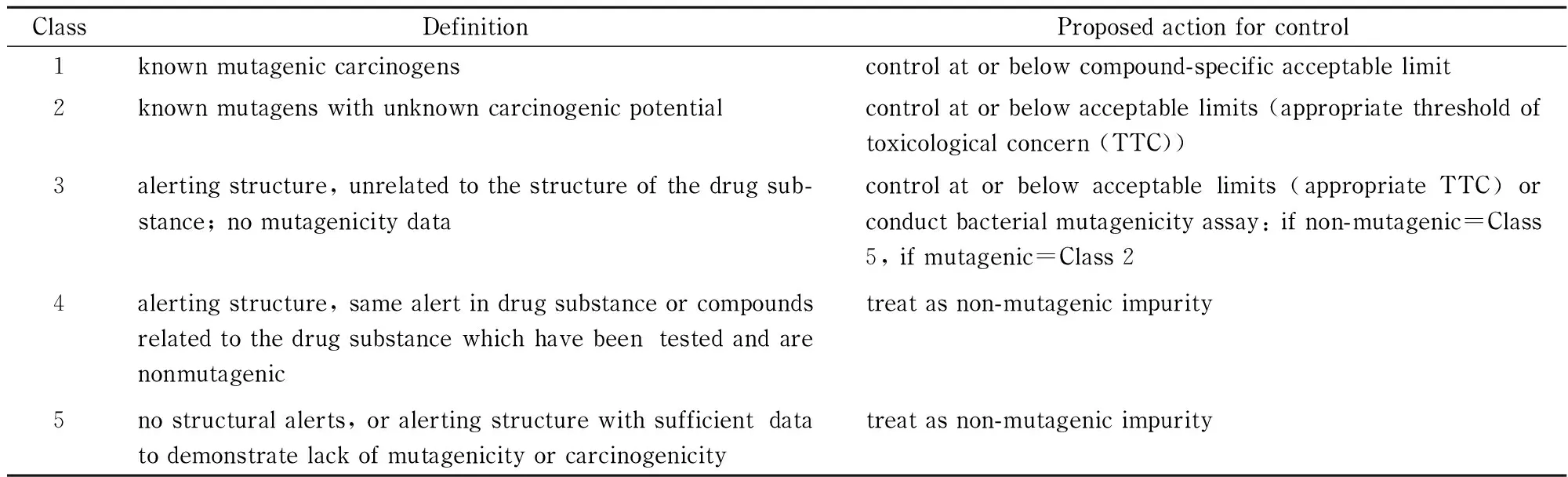
表 1 按诱变性和潜在致癌性的杂质分类及相应的控制措施
EMA人用药物委员会安全工作组就之前的《基因毒性杂质限度指南》发布了相关的问答[10],该问答接纳了Müeller等的提议,采用“阶段化TTC”的方法来确立临床研发中具有不同用药周期要求的药物中基因毒性杂质的可接受摄入量。此外,该问答中回应了对“ALARP”原则的质疑,表示如果药物中的基因毒性杂质经考察已经低于规定的限度,则无需采用“ALARP”原则继续降低药物中基因毒性杂质的残留量,由此有效降低了分析方法建立或者转移带来的挑战。美国食品药品监督管理局(U. S. Food and Drug Administration, FDA)[11]于2008年12月也出台了有关原料药和制剂中基因毒性杂质控制的指南草案(Guidance for industry-genotoxic and carcinogenic impurities in drug substances and products: recommended approaches)。该草案与EMA颁布的限度指南在服药周期超过1个月至终生服药时所涉及的TTC限度要求完全一致,而在服药周期短于1个月的具体阶段化TTC值规定则有所不同。ICH (the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use)[12]于2014年7月15日正式发布了基因毒性杂质研究指南(M7),并在多次修改后于2017年5月31日出台了第四版指南M7(R1),进一步肯定了上述各方采用“阶段化TTC”以及“五分类”策略对基因毒性杂质进行研究和控制的科学性,并在这些策略的基础上做了一定的调整和增补。
表2对比显示了各监管机构及PhRMA对于基因毒性杂质的控制要求。可以看到EMA和FDA的规定最为细致,且两者对于具有1个月以上服药周期的药物监管要求完全一致,而ICH的监管要求相对而言较为宽松。在同样12个月的服药周期内,EMA和FDA都规定了每天不得超过5 μg的限度,而ICH的要求放宽到20 μg。在服药周期12月以上时,PhRMA、EMA和FDA规定基因毒性杂质每天的可接受摄入量不得过1.5 μg,而ICH则要求只有服药周期10年以上时,才需限定每日摄入的基因毒性杂质不得超过1.5 μg。究其原因,可能是由于ICH作为多方协调机构,兼顾了多国的技术可行性。

PhRMA: pharmaceutical research and manufacturers of America; EMA: European Medicines Agency; FDA: U. S. Food and Drug Administration; ICH: the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.
针对药物中存在多个诱变性杂质的情况,M7(R1)也提出了相应的监管要求。当药物中含有多个1类杂质时,应根据每个杂质的特定限度分别对其进行控制。当药物中含有两个2类或3类杂质时,每个杂质的控制仍然按照“阶段化TTC”的原则控制,即药物中的每个杂质限度必须满足表2的规定;然而,如果药物中存在3个及以上2类或3类杂质,每个杂质的可接受摄入量在满足表2所述限度的同时,所有杂质的总量还需要根据表3进行限定。

与欧美发达国家相比,我国基因毒性杂质的研究尚处于起步阶段,目前也没有直接出台相关的指南,但是在当下药物研发全球化趋势下,特别自2017年6月我国加入ICH组织以来,我国药监部门主要是参照上述国际主流指南开展药物中的基因毒性杂质的审批与监管,以保证药物质量的可控性与临床应用的安全性。
2 磺酸酯类基因毒性杂质研究进展
由于药物与磺酸成盐后具有溶解度增加、溶解速率加快、稳定性增强、引湿性降低以及晶型更稳定等优点,且磺酸盐药物在改善有机弱碱性药物的机械性质,诸如流动性、黏合性、可压缩性等方面也具有独特的优势,因此通过与磺酸成盐是一种提高有机弱碱药物成药性的重要手段[13]。据统计,目前处于临床研发阶段和已经上市的磺酸盐药物有两百余种(见表4),治疗病症包括呕吐、哮喘、血栓、细菌感染、中风、癌症、艾滋病、抑郁症等类型[14],由此可见磺酸盐药物的开发和市场应用具有一定的普遍性和广泛性。磺酸盐药物由磺酸和药物自由碱通过成盐反应得来,在合成过程中用到低级醇类溶剂(甲醇、乙醇、丙醇和异丙醇等)时,磺酸就可能与这些醇类溶剂发生副反应产生磺酸酯杂质。早在2000年,欧洲药品质量管理局(European Directorate for the Quality of Medicines, EDQM)即要求密切关注磺酸盐药物在成盐过程中形成基因毒性杂质磺酸酯的潜在风险,并且要求除了要对甲磺酸盐中的烷基磺酸酯进行限度检查外,还要提供其他相关的研究信息[15]。在这之后发生的甲磺酸奈非那韦因甲磺酸乙酯超标而遭到撤市的事件[16],更是成为触发欧洲药监部门和制药企业高度关注磺酸酯基因毒性的标志性事件。Roche公司通过对生产工艺的调查,发现甲磺酸乙酯残留超标的原因是甲磺酸贮存罐清洗所用的乙醇未被完全清除,以较高的浓度水平残留在贮存罐中,与存放其中的甲磺酸反应生成了甲磺酸乙酯。在Roche公司解决了甲磺酸乙酯污染问题并对其毒性进行评估后,欧洲药品评价局才恢复了甲磺酸奈非那韦在欧洲的市场授权。该事件的发生不仅极大地危害了病人的健康,也给Roche公司造成了巨大的经济损失。

/: not reported.
除了磺酸和醇可能生成磺酸酯以外,磺酸的衍生物在与醇类溶剂共存时也可能会发生副反应从而产生磺酸酯。如磺酰氯、磺酸酐等,其化学性质活泼,在药物合成中具有非常广泛的用途,可作为良好的离去基团、某个化学结构基团的保护基团等参与化学反应;而且这些衍生物在重要的药物合成前体——磺酰胺类化合物的合成中以及手性异构体药物的分离中也起着非常重要的作用,在这些化学反应中,假如同时使用醇溶剂,就可能会发生副反应而产生磺酸酯[17](见图2)。

图 2 磺酸、磺酸卤化物、磺酸酐与低相对分子质量醇反应生成磺酸酯示意图Fig. 2 Schematic generation of sulfonate esters between sulfonic derivatives and low relative molecular mass alcohols X=OH, Cl, Br, I, OSO2R; R=alkyl, phenyl; R′=alkyl.
由于醇类溶剂和磺酸及其衍生物的种类多样,因此由两者生成的磺酸酯包含多种结构,从最简单的烷基磺酸酯到复杂的芳基磺酸酯,主要有:甲磺酸酯,如甲磺酸甲酯、乙酯、丙酯、异丙酯;苯磺酸酯,如苯磺酸甲酯、乙酯、丙酯、异丙酯;对甲苯磺酸酯,如对甲苯磺酸甲酯、乙酯、丙酯、异丙酯;三氟甲磺酸酯,如三氟甲磺酸甲酯、乙酯、丙酯、异丙酯等(见图3)。研究表明,磺酸酯具有诱变性,能够直接或者经代谢活化后间接地将自身结构上的烷基残基转移到富电子的DNA碱基上而引起DNA的烷基化,从而导致遗传物质的损伤[18,19]。
2.1 磺酸酯的形成机理及控制策略

图 3 磺酸酯的种类Fig. 3 Categories of sulfonate esters
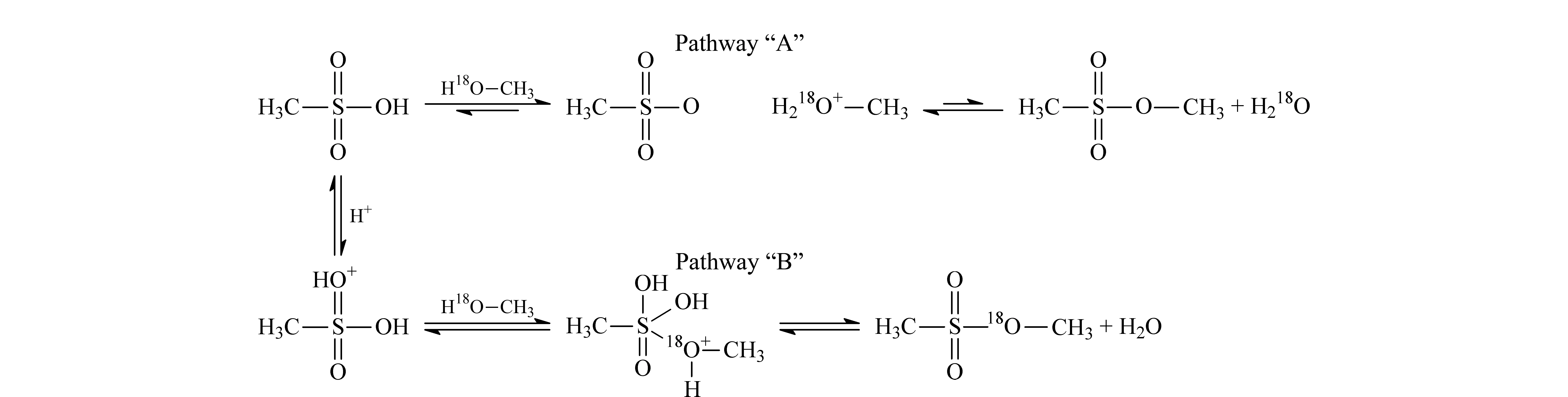
图 4 甲磺酸和甲醇反应机理[20]Fig. 4 Reaction mechanism between methanesulfonic acid and methanol[20]
由葛兰素史克、阿斯利康等药企研发人员组成的药物质量研究所工作组在Teasdale领导下,利用同位素(18O)标记的方法,探究了甲磺酸和甲醇反应生成甲磺酸甲酯的机理[20]。起初Teasdale等推测甲磺酸和甲醇的反应机理有两种可能途径(见图4),然而同位素测定结果显示只在产物水中检测到了18O,而产物甲磺酸酯中并不存在18O,说明甲磺酸和甲醇反应生成甲磺酸甲酯的机理为图4中所示的途径A,即首先甲磺酸释放质子,形成甲磺酸根离子,同时甲醇质子化形成氧鎓离子,然后甲磺酸根离子对该氧鎓离子进行亲核进攻,生成甲磺酸甲酯和水。该反应机理揭示了磺酸和醇反应生成磺酸酯需要一定的酸性环境。接着Teasdale等[21]又继续探讨了反应温度,体系中的水分含量,溶液酸度等因素对磺酸酯生成和消除的影响,结果表明降低反应温度能显著降低磺酸酯形成的速率和相应产生的磺酸酯的水平;缩短醇和甲磺酸的反应时间、增加水分的含量、增大反应体系的pH也能够有效减少磺酸酯的生成。他们的研究提供了尽可能降低磺酸酯水平的控制策略[22]:在磺酸盐药物成盐过程中,尽量按照化学计量比投加磺酸和自由碱,同时为了促进向成盐反应的正向进行,采取加入过量自由碱而非磺酸的方式使反应体系呈弱碱性,从而消除磺酸酯生成的风险;如果必须要使用过量的磺酸,应尽可能减少其用量并在较低的温度下进行成盐反应以及后续的分离操作,降低磺酸酯生成的速率;减少成盐反应和重结晶所用的时间,以此减少磺酸和醇类溶剂共存发生反应的时间;向成盐反应和后续分离步骤所用的溶液中添加适量的水,减少质子化醇的浓度,使酯化平衡反应向逆反应方向移动以加速磺酸酯的水解;避免磺酸和醇在使用前预先混合和储存的情况。这些策略为磺酸盐药物合成中磺酸酯的有效控制提供了重要参考,当然要注意到这些措施可能对反应收率带来负面影响,需要在权衡各控制措施对药物合成带来的利弊基础上进行合理选择与优化。
2.2 磺酸酯相关分析方法
为了确保药物的质量安全,有必要建立准确、耐用的分析检测方法,对药物中残留的磺酸酯进行定量测定并及时与合成工艺人员进行反馈交流,以确保工艺路线的合理安全。目前文献[23-43]报道的磺酸酯的分析方法主要有液相色谱(HPLC)法和气相色谱(GC)法,相关方法的回收率和检出限总结于表5中。
2.2.1液相色谱法
非挥发性的磺酸酯可采用HPLC进行分析,其中具有紫外吸收的磺酸酯的检测可采用紫外(UV)检测器。为了提高检测灵敏度,常常会进一步选用选择性和灵敏度更高的质谱(MS)检测器。Zheng等[23]利用LC/UV法检测奥格列汀原料药中的苯磺酸异丙酯,以220 nm作为测定波长,以添加0.1% (体积分数)乙酸的水/乙腈(65∶35, v/v)作为流动相。该方法的检出限和定量限分别为0.33 mg/L 和1 mg/L。由于磺酸酯具有遇水易分解的特性,因此在采用LC法进行磺酸酯测定时,需要特别关注磺酸酯的溶液稳定性[24-26],否则可能导致假阴性的检测结果。Kakadiya等[27]采用电喷雾电离(ESI)源,建立了LC/MS/MS法分别检测洛匹那韦和利托那韦原料药中的甲磺酸甲酯和甲磺酸乙酯,检出限约为2 μg/L,回收率在80% ~120%之间。陈成等[28]则采用了大气压化学电离(APCI)源检测2-脱氧-D-核糖中甲磺酸甲酯和甲磺酸乙酯,该方法的检出限为4 μg/L,回收率为91.7% ~113.1% 。Guo等[29]进一步对APCI和ESI这两种电离源进行了对比,结果发现采用ESI时,方法的灵敏度和重现性较差。相较而言,采用APCI电离方式的负离子模式,各待检磺酸酯的灵敏度较高,因此作者最终选用APCI方式用于甲磺酸伊马替尼和长春西汀原料药中磺酸酯的检测,方法灵敏度在2 μg/L 左右,回收率在93% ~118%之间。针对复杂药物基质,采用液液萃取等样品前处理方式可以减少辅料等带来的干扰。Cappiello等[30]在检测对乙酰氨基酚片中的对甲苯磺酸甲酯等烷基化试剂时,采用四氢呋喃作为萃取溶剂,然后采用电子轰击(EI)电离源的LC/MS法进行检测,回收率在55% ~82%之间,可以看出该方法的准确性波动较大。

表 5 文献报道的磺酸酯检测方法的检出限及回收率汇总
IBS: isopropyl benzenesulfonate; MBS: methyl benzenesulfonate; EBS: ethyl benzenesulfonate; BBS: butyl benzenesulfonate; MTS: methylp-toluenesulfonate; ETS: ethylp-toluenesulfonate; ITS: isopropyl p-toluenesulfonate; MMS: methyl methanesulfonate; EMS: ethyl methanesulfonate; PMS: propyl methanesulfonate; IMS: isopropyl methanesulfonate; PBS: propyl benzenesulfonate; PTS: propylp-toluenesulfonate; DMS: dimethyl sulfate; DES: diethyl sulfate; DPS: dipropyl sulfate; DIPS: diisopropyl sulfate; API: active phamaceutical ingredient; SM934: an artemisinin derivative.
由于磺酸酯属于酯类结构,在含水的流动相中可能会发生水解等问题而影响测定结果的准确性,因此利用化学衍生化的方式将其转化为稳定的物质是解决磺酸酯在线水解问题的一种较好的方式。An等[31]建立了衍生化LC/MS法检测甲磺酸甲酯、甲磺酸乙酯、甲磺酸丙酯、甲磺酸异丙酯、苯磺酸甲酯、苯磺酸乙酯、苯磺酸丙酯、苯磺酸异丙酯、对甲苯磺酸甲酯、对甲苯磺酸丙酯、对甲苯磺酸异丙酯、硫酸二甲酯、硫酸二乙酯、硫酸二丙酯和硫酸二异丙酯等多种烷基酯类基因毒性杂质,采用10%(体积分数)三甲胺水溶液作为乙酯、丙酯和异丙酯的衍生化试剂,10%(体积分数)三乙胺水溶液作为甲酯的衍生化试剂,在50~60 ℃条件下孵育60 min,衍生为较稳定的季铵盐,采用HILIC(hydrophilic interaction chromatography)亲水色谱柱进行方法学的开发和验证。但是该衍生化方法所得到的衍生产物的色谱峰出现了较为明显的拖尾现象,而且该方法对于不同待测物的准确性有比较大的差异,表现为回收率跨度较大,如苯磺酸丙酯的回收率为85% ,而苯磺酸乙酯的回收率高达137% 。Zhou等[32]则利用N,N-二乙基二硫代氨基甲酸作为衍生化试剂,向待测甲磺酸甲酯和乙酯中引入生色基团进行柱前衍生,采用UV检测器进行检测。但该衍生化时间较为冗长,需耗时60 min。
2.2.2气相色谱法
为了避免在采用LC法检测磺酸酯时出现遇水易分解现象,具有一定挥发性或者半挥发性的磺酸酯可以采用GC进行检测,并采用氢火焰离子化检测器(FID)或者MS检测器。Li[33]和郑飞等[34]均采用直接进样GC/FID法检测原料药中的甲磺酸甲酯、乙酯和异丙酯,进样量为5 μL,进样口温度为120 ℃。该方法由于引入大量的非挥发性药物基质,容易污染进样口,因此需要及时更换衬管。此外,该方法由于采用FID检测器,灵敏度较低,仅能检测40 μg/L 及以上的磺酸酯。Ramakrishna等[35]报道了采用直接进样GC/MS法来测定甲磺酸伊马替尼中的甲磺酸甲酯和乙酯,以正己烷作为样品溶剂,采用DB-1色谱柱(30 m×0.25 mm×0.25 μm),进样口温度设定为140 ℃,进样量为2 μL,分流比为200∶1,其检出限和定量限分别为0.3和1.0 mg/L,在1~15 mg/L 范围内的回收率为98.1% ~99.6% 。Zhang等[36]也报道了直接进样GC/MS法来对甲磺酸伊马替尼中的甲磺酸甲酯和甲磺酸乙酯进行定量分析。与上述Ramakrishna等建立的方法不同,该方法采用水饱和的正己烷作为样品溶剂,以HP-5MS(30 m×0.32 mm×0.25 μm)为色谱柱,得到的灵敏度更高,检出限为1 μg/L,定量限为5 μg/L,在10~1 000 μg/L 范围内的准确度为97.2% ~99.8% 。该方法同时还提高了进样口温度(200 ℃),分流比也减小到100∶1。通过对比上述Zhang等与Ramakrishna等的研究工作,提示了样品溶剂、进样口温度和分流比等条件的优化对于提高磺酸酯检测方法的灵敏度至关重要。
虽然直接进样操作简单,但由于会因此引入大量非挥发性基质而导致色谱系统污染从而出现平行性异常等问题,研究人员尝试了固相微萃取(SPME)、液液萃取(LLE)作为样品前处理方式的GC/MS法来检测药物中的磺酸酯。Colón等[37]建立了直接浸入式SPME-GC/MS的方法对药物中的甲磺酸甲酯、甲磺酸乙酯、苯磺酸甲酯、苯磺酸乙酯、对甲苯磺酸甲酯、对甲苯磺酸乙酯进行限度检查,采用弱极性的聚二甲基硅氧烷/二乙烯基苯(PDMS/DVB)为固定相的微萃取头,以20 mmol/L 磷酸盐缓冲液(pH 4.7)作为样品溶剂,在该pH条件下药物基质被离子化而增大了极性,不易被固定相萃取从而减小了基质效应。Wollein等[38]采用LLE的样品前处理方式检测药物固体制剂中的甲磺酸甲酯、乙酯、丙酯以及苯磺酸甲酯和乙酯,测得检出限和定量限分别小于5和20 μg/L。由于该方法采用非极性的正己烷作为萃取溶剂,因此不适用于极性较大的药物中磺酸酯的测定。
尽管SPME、LLE能在一定程度上降低基质效应,但操作繁琐,耗时耗力而且磺酸酯在进样口和衬管中的热降解问题并没有得到解决,因此,研究人员进一步采用顶空衍生化的方式,将反应性的磺酸酯衍生为较为稳定的、挥发性的物质。Lee等[39]采用硫氰酸钠作为衍生化试剂,以水为溶剂,建立了顶空GC/MS方法检测一种甲磺酸盐中的甲磺酸甲酯、乙酯和异丙酯。然而该衍生方法会产生异构化的产物(烷基硫氰酸和烷基异硫氰酸),从而不利于进行定量测定,而且该方法仅仅局限于水溶性药物,不适用于水溶性较差的药物中磺酸酯的检测。Alzaga等[40]采用五氟苯硫酚作为衍生化试剂(其溶剂为水和二甲亚砜混合溶液),将磺酸酯衍生为五氟苯硫醚,采用顶空GC/MS检测。在方法验证的过程中发现,该方法的准确性(尤其是对甲苯磺酸甲酯)会明显受到基质的影响,检测样品A中的对甲苯磺酸甲酯的回收率仅有40% ,而对甲苯磺酸乙酯和异丙酯的回收率均大于93% ;样品B中所有酯的回收率则在85%和100%之间。欧洲药典(EP)[41]也描述了一种在线衍生化顶空GC/MS法检测甲磺酸倍他司汀、苯磺酸氨氯地平和对甲苯磺酸舒他西林原料药中的痕量甲磺酸酯、苯磺酸酯和对甲苯磺酸酯。采用碘化钠(NaI)作为衍生化试剂,利用碘离子的亲核性,将磺酸酯在线衍生为不同种类的碘代烷烃,经GC/MS进行分析检测。该方法因衍生化反应操作简便,具有较好的通适性而受到分析人员的青睐。范达等[42]采用了该衍生化方法检测注射用甲磺酸吉米沙星中的甲磺酸甲酯、乙酯、异丙酯,以电子捕获检测器(ECD)代替了MS检测器。发现该方法对于不同磺酸酯的检测灵敏度差异较大,其中甲磺酸甲酯的灵敏度最高,检出限为0.002 μg/L,甲磺酸异丙酯的灵敏度最低,检出限为0.25 μg/L。Liu等[43]发现EP采用的用于防止NaI氧化的抗氧剂硫代硫酸钠对方法的耐用性具有不利影响,在延长顶空平衡时间和升高平衡温度时会造成对甲苯磺酸甲酯的衍生产物碘甲烷(MeI)的降解。研究发现选用维生素C作为替代抗氧剂,能够在较高的平衡温度和较长的平衡时间下保持包括MeI在内的衍生产物的稳定性,并提高了该方法的耐用性并扩展了相应的应用范围,检测限和定量限可以分别达到0.6和2 μg/L。
3 总结与展望
近年来有关药物中的基因毒性杂质的控制得到来自于药物监管部门与药物研发机构的高度重视,基于风险管理的“阶段化TTC”控制策略,使得基因毒性杂质的控制更趋科学合理。磺酸酯作为一类较为常见的基因毒性杂质,可以通过对关键工艺参数的优化与设计进行有效控制,符合当下“质量源于设计”的理念,即良好的工艺设计往往能促进高质量药物的生产,并在充分保障药物安全性的基础上尽可能减少企业的研发投入及相应的政府监管成本。
由于基因毒性杂质在药物中的控制较为严格,通常在μg/L甚至接近ng/L水平,因此需要建立高灵敏度的定量检测方法,其中需要综合考虑药物基质以及待测磺酸酯的挥发性、热稳定性以及化学反应特性等,有的放矢地建立满足灵敏度需求的检测方法。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
中国民间疗法(2021年17期)2021-11-04
当代医药论丛(2017年22期)2017-04-12
国外医药(抗生素分册)(2016年2期)2016-07-12
中国卫生标准管理(2015年4期)2016-01-14
淮海医药(2015年2期)2016-01-12
中国生化药物杂志(2015年4期)2015-07-07
中国当代医药(2015年9期)2015-03-01
湖南师范大学自然科学学报(2015年2期)2015-02-27
中华皮肤科杂志(2014年4期)2014-12-19
