
LPS联合z-VAD-FMK介导IFN-γ预处理巨噬细胞发生程序性坏死*
2018-09-27 11:17章述军黄文祥
中国病理生理杂志 2018年9期
章述军, 肖 晴, 黄文祥
(重庆医科大学附属第一医院感染科, 重庆市传染病与寄生虫重点实验室, 重庆 400016)
巨噬细胞是天然免疫的重要成员之一,在感染、自身免疫疾病和肿瘤的发生发展中均起着十分重要的作用,根据诱导其分化的细胞因子的种类和细胞功能的不同,主要包括M1和M2亚型。M1亚型巨噬细胞主要由Th1类细胞因子如干扰素γ(interfe-ron-γ,IFN-γ)等诱导分化而来,通过分泌肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白细胞介素1β(interleukin-1β,IL-1β)和IL-6等炎症因子而促进炎症反应; 而M2亚型主要由Th2类细胞因子如IL-4和IL-13等诱导分化而来,通过分泌IL-10和转化生长因子β (transforming growth factor-β,TGF-β)等抑制炎症、促进组织修复,两者间的平衡是维持稳态的重要保障[1]。在感染的早期以巨噬细胞以M1亚型为主,通过促进炎症反应清除病原体,在感染后期以M2亚型为主,以促进炎症消除,修复组织,但目前这种转换的机制仍不清楚[2]。
细菌脂多糖(lipopolysaccharide,LPS)是细菌重要的病原相关分子模式之一,是诱导巨噬细胞向M1亚型分化、促进炎症反应的重要因子。在细菌感染和严重肝脏疾病时,LPS也是重要的致病因子,除引发M1亚型巨噬细胞炎症应答外,还同时促进非M1亚型的巨噬细胞向M1亚型巨噬细胞进行分化,进一步加强机体的炎症状态,如果不加控制,机体会出现严重的免疫过激状态导致多器官功能损伤而死亡。因此,机体存在相应的保护机制来限制炎症的进展,LPS可能参与其中[3]。
因此,本研究采用THP-1细胞体外模拟巨噬细胞及其M1亚型,探讨LPS是否对其死亡具有影响,及其可能机制。
材 料 和 方 法
1 细胞及培养
人THP-1细胞系(TIB-2020)和胎牛血清(30-2020)购自ATCC;RPMI-1640培养基 (11835055)、β-巯基乙醇(31350010)、重组人IFN-γ(PHC4031)购自Gibco;佛波酯(phorbol 12-myristate 13-acetate,PMA;P1585)购自Sigma;z-VAD-FMK(S7023)、necrostatin-1(S8037)、VX765(S2228)、GSK872(S8465)和SP600125(S1460)购自Selleck。用于实验的THP-1细胞不超过20代, 生长密度不超过1×109/L,细胞培养于含10%胎牛血清和50 μmol/L巯基乙醇的RPMI-1640培养基,置于37 ℃、5% CO2培养箱中。以PMA(100 μg/L, 160 nmol/L)刺激24 h后以无菌磷酸缓冲盐溶液(phosphate-buffered saline,PBS)淋洗2次,再更换为不含PMA的完全培养基继续培养48 h。对于PMA诱导后的巨噬细胞,本文将其作为M0巨噬细胞, 继以IFN-γ (50 μg/L)刺激24 h诱导分化后作为M1亚型细胞,相关基因谱的检测与鉴定详见既往报道[4]。
2 主要方法
2.1细胞死亡的检测 本研究中,我们采用乳酸脱氢酶(lactate dehydrogenase,LDH)释放实验和末端脱氧核苷酸转移酶介导的dUTP缺口末端标记(terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling,TUNEL)法进行细胞死亡的判定。LDH释放检测试剂盒(88954)购自于Thermo Fisher Scientific,严格按照说明书进行实验:接种0.5×109/L的THP-1细胞于培养板,诱导分化M0和M1亚型巨噬细胞,给予LPS(100 μg/L)分别刺激6、12和24 h后,收集上清检测释放出的LDH,裂解剩余的贴壁细胞检测LDH,两者之和即为总LDH,上清中的LDH除以总LDH即为所释放的LDH。TUNEL试剂盒(G3250)购自于Promega,处理后的细胞先以PBS淋洗2遍,后以4%多聚甲醛-PBS溶液于室温固定10 min,随后按说明书操作。随机取5个视野计数阳性信号细胞,对比4’,6-二脒基-2-苯基吲哚(4’,6-diamidino-2-phenylindole,DAPI)染色计算阳性率。
2.2Western blot实验 巨噬细胞接受相应的处理后,以无菌冷PBS淋洗2遍,加入RIPA裂解液后刮取收集于EP管,继续于冰上裂解30 min,期间采用超声破碎3次,每次5 s。14 000×g离心15 min后保留上清,测定蛋白浓度后取40 μg每泳道以90 V电泳90 min,90 V下转膜120 min,5%脱脂奶粉-PBST溶液封闭1 h,然后与相应 I 抗于4 ℃孵育过夜(16 h),PBST溶液漂洗3次,每次5 min,与 II抗于室温下孵育1 h,PBST漂洗3次,每次5 min,后使用ECL液发光于ChemiDoc XRS+系统(Bio-Rad)曝光采集图像,使用ImageJ软件分析。所采用的抗体:GAPDH(内参照)抗体(sc-367714)购自Santa Cruz;受体相互作用蛋白3(receptor-interacting protein 3,RIP3)抗体(ab56164)购自Abcam;RIP1抗体(4926)、总c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)抗体(9252)、磷酸化JNK抗体(9251)、总细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)抗体(9102)、磷酸化ERK抗体(9101)、总P38抗体(9212)、磷酸化P38抗体(9211)和NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)抗体(15101)均购自Cell Signaling Technology。
3 统计学处理
使用SPSS 22.0进行统计分析。所有实验均独立完成3次, 计量资料以均数±标准差(mean±SD)表示。两组间比较采用Student’st检验,多组间比较进行正态分布性检验和方差齐性检验后采用单因素方差分析(one-way ANOVA),多组间两两比较采用SNK-q检验,以P<0.05为差异有统计学意义。
结 果
1 LPS联合z-VAD-FMK特异性诱导IFN-γ预处理巨噬细胞死亡
分化后的M0和M1亚型巨噬细胞接受LPS(100 μg/L)刺激后检测LDH释放和TUNEL阳性率。在接受LPS刺激0 h、6 h、12 h和24 h后,M0亚型巨噬细胞LDH释放依次为(7.1±0.0)%、(9.1±0.6)%、(12.0±1.0%)和(12.8±0.7)%,而M1亚型依次为(8.2±0.0)%、(16.8±0.6)%、(22.3±1.2)%和(22.4±0.8)%,见图1A。TUNEL阳性率在LPS刺激0 h、6 h、12 h和24 h时在M0亚型依次为(0.3±0.1)%、(4.8±0.2)%、(3.8±0.5)%和(2.3±0.4)%;M1亚型依次为(1.0±0.2)%、(12.9±1.0)%、(11.2±1.0)%和(8.5±1.7)%,见图1B。2个亚型的巨噬细胞接受LPS刺激后其LDH 释放和TUNEL阳性率均显著升高(P<0.05),除基线(0 h)外,LPS处理6 h、12 h和24 h后M1亚型巨噬细胞的LDH释放及TUNEL阳性率均显著高于M0亚型(P<0.05)。
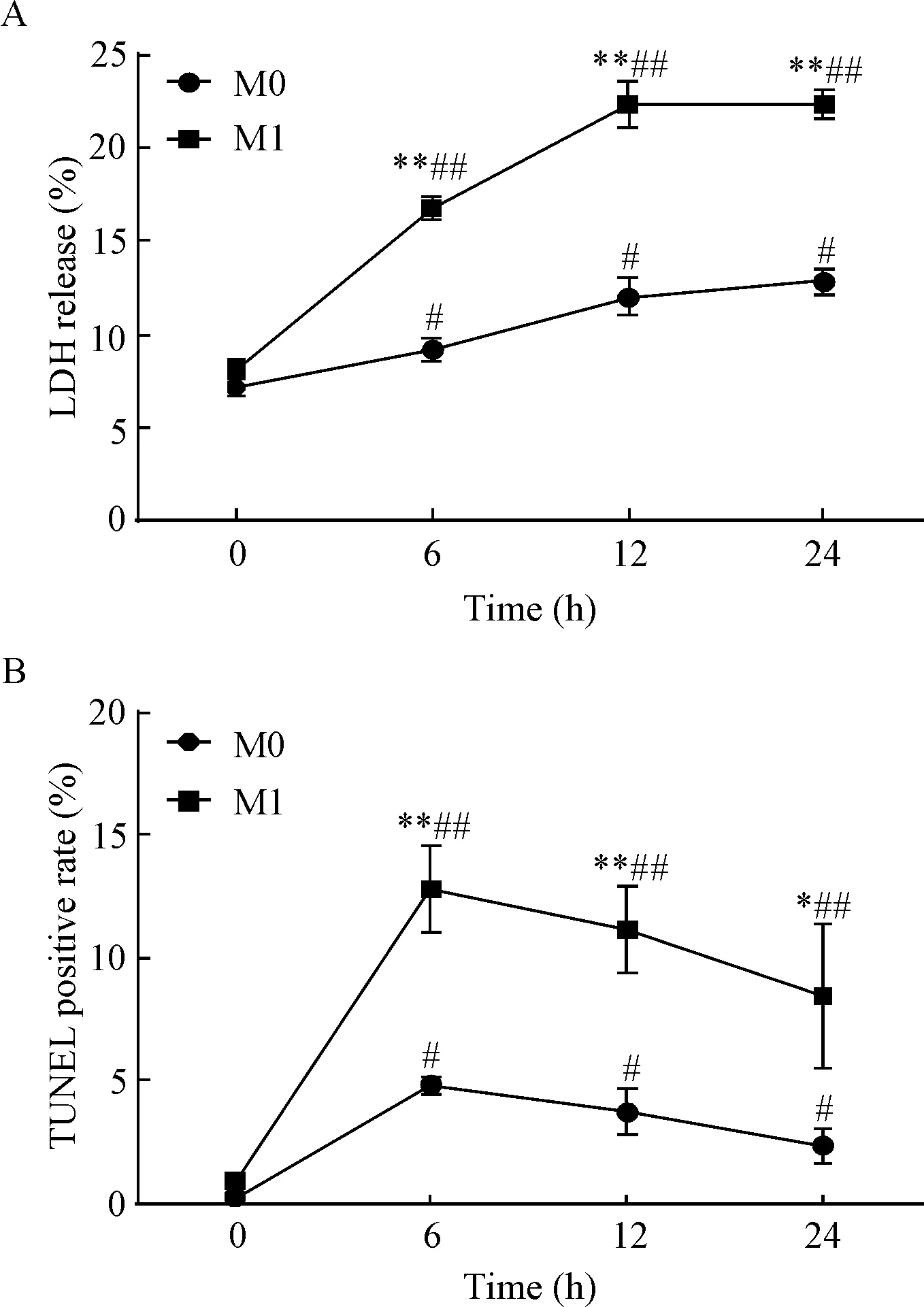
Figure 1. LPS at 100 μg/L induced significantly higher LDH release (A) and TUNEL positive rate (B) in M1 macrophages than M0 macrophages. Mean±SD.n=3.*P<0.05,**P<0.01vsM0;#P<0.05,##P<0.01vs0 h.
图1LPS对M0和M1亚型巨噬细胞死亡的影响
LPS处理2种亚型巨噬细胞后,LDH释放在12 h达峰值,TUNEL阳性率在6 h达峰值,因此,对于LDH释放和TUNEL阳性率分别选择12 h和6 h作为时间节点,进一步使用泛caspase抑制剂、caspase-1抑制剂和RIP1抑制剂以观察对细胞死亡的影响并分析其死亡模式。结果显示,LPS介导的LDH释放和TUNEL阳性率在M0细胞均可被泛caspase抑制剂z-VAD-FMK(20 μmol/L)阻断;在M1亚型细胞,z-VAD-FMK(20 μmol/L)阻断了TUNEL阳性率的升高(P<0.01),但显著增强了LDH释放(P<0.01)。而caspase-1 抑制剂VX765(20 μmol/L)及RIP1抑制剂necrostatin-1(100 μmol/L)对LPS介导的M1亚型巨噬细胞的LDH释放和TUNEL阳性率均无影响。而LPS联合z-VAD-FMK所介导的LDH释放可以被necrostatin-1所阻断,见图2。因此,LPS介导的M1亚型巨噬细胞凋亡,加用z-VAD-FMK后TUNEL阳性率被抑制,LDH释放增加,而升高的LDH释放可被necrostatin-1完全阻断,考虑LPS联合z-VAD-FMK诱导M1亚型巨噬细胞发生了程序性坏死,而LPS联合z-VAD-FMK对M0巨噬细胞并无此作用。
2 IFN-γ上调巨噬细胞RIP3的表达
上述结果提示LPS联合z-VAD-FMK可特异性诱导M1巨噬细胞发生程序性坏死,可能是一种限制炎症反应的自我保护机制。为进一步探讨LPS联合z-VAD-FMK特异性发生于M1亚型巨噬细胞而非M0亚型的机制,我们比较了M0和M1亚型巨噬细胞RIP1和RIP3的表达,与M0亚型巨噬细胞相比较,IFN-γ 能够显著上调M1亚型巨噬细胞RIP3的表达(P<0.05),后续的LPS刺激使得其进一步升高,而无论IFN-γ还是LPS都不能在巨噬细胞中上调RIP1的表达,见图3。
3 IFN-γ增强LPS介导的巨噬细胞JNK磷酸化和NLRP3的表达
除了RIP1和RIP3是参与程序性坏死的重要分子外,LPS还可通过结合巨噬细胞表面的Toll样受体4(Toll-like receptor 4,TLR4)而进一步激活丝裂原激活蛋白激酶(mitogen-activated protein kinase,MAPK)通路,其中JNK的活化也与程序性坏死的发生密切相关。因此我们首先检测了IFN-γ对MAPK通路的活化的影响,发现LPS可诱导P38和JNK磷酸化(P<0.05),而IFN-γ 能够增强LPS所诱导的JNK磷酸化(P<0.05),而对LPS介导的P38磷酸化无显著影响,见图4。既往有报道JNK的活化需要NLRP3的参与[5],因此本研究也检测了IFN-γ对NLRP3表达的影响,结果发现IFN-γ能显著上调NLRP3的表达(P<0.05),见图5。
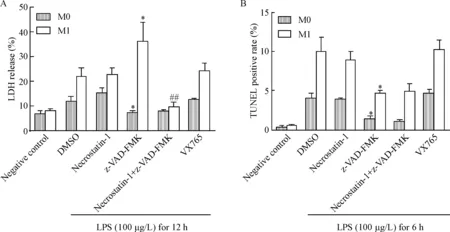
Figure 2. The effects of various inhibitors on LPS-mediated LDH release (A) and TUNEL positive rate (B) of macrophages. Mean±SD.n=3.*P<0.05vsDMSO group;##P<0.01vsz-VAD-FMK group.
图2不同抑制剂对LPS介导的巨噬细胞LDH释放和TUNEL阳性率的影响
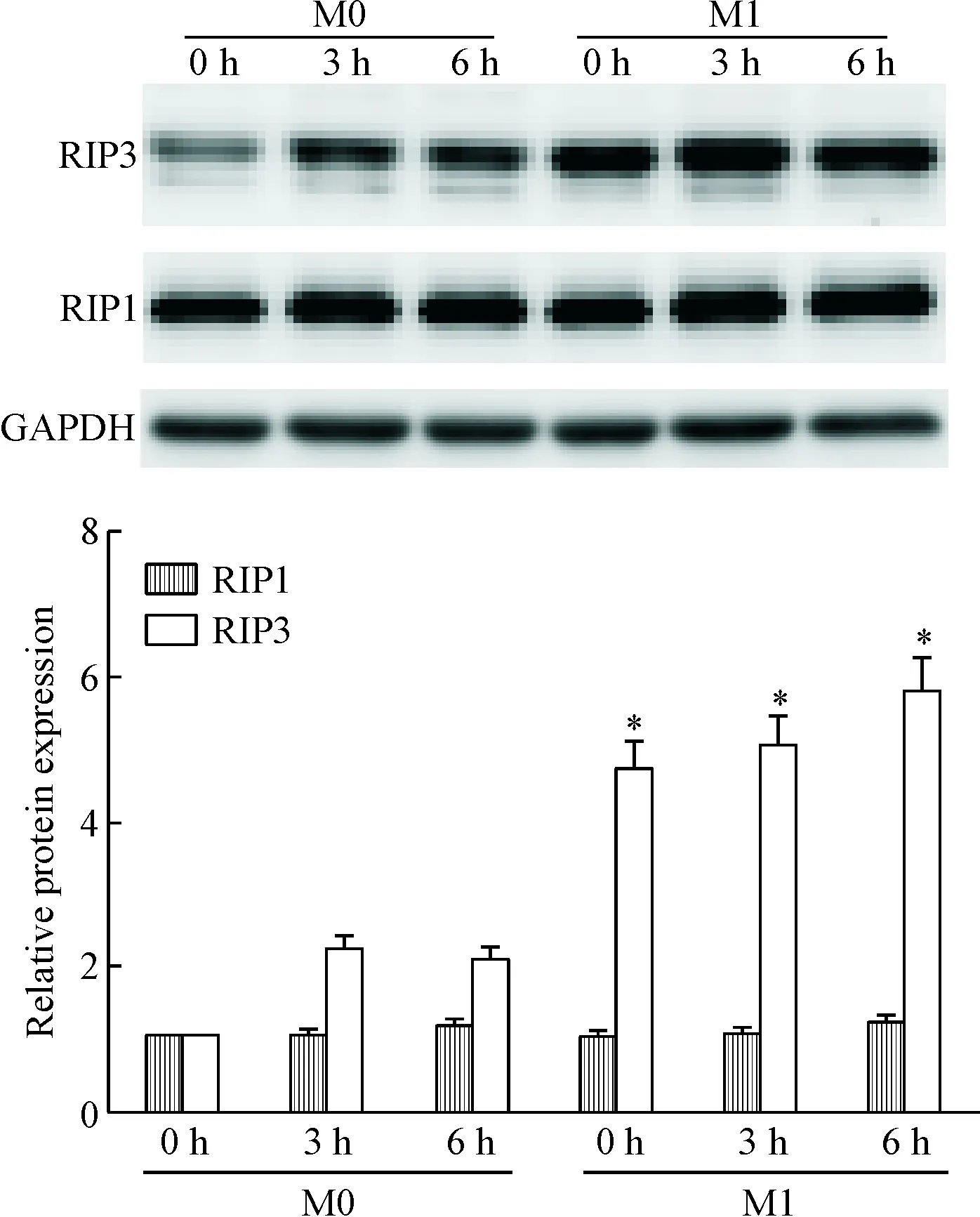
Figure 3. INF-γ upreguated the expression of RIP3, but not RIP1, in the macrophages. Mean±SD.n=3.*P<0.05vsM0.
图3IFN-γ上调巨噬细胞RIP3的表达而对RIP1无影响
4 GSK872和SP600125可阻断LPS联合z-VAD-FMK介导的IFN-γ预处理巨噬细胞死亡
上述结果提示IFN-γ可能是通过上调RIP3和促进JNK磷酸化,使得巨噬细胞在接受LPS联合z-VAD-FMK处理时发生程序性坏死。为进一步明确RIP3和JNK的作用,进一步探讨了RIP3抑制剂GSK872(5 μmol/L)和JNK抑制剂SP600125(10 μmol/L)对IFN-γ预处理的巨噬细胞在LPS联合z-VAD-FMK介导的程序性坏死中的作用。如图6所示,GSK872和SP600125均可显著阻断LDH的释放(P<0.05)。
讨 论
巨噬细胞在感染早期以M1亚型为主,促进炎症反应从而清除病原体,后期以M2亚型为主,促进炎症的消除和组织修复。既往文献报道在酒精性肝炎小鼠模型中, M2亚型巨噬细胞可介导M1亚型巨噬细胞发生凋亡,从而限制炎症反应,促进组织修复[6]。目前认为,对于没有进一步分化的M0巨噬细胞,LPS在激活凋亡通路的同时也会激活促进增殖的核因子κB(nuclear factor-kappa B,NF-κB)和ERK通路从而维持细胞存活。但在某些情况下,LPS也可直接诱导巨噬细胞通过多种方式死亡,例如在NF-κB通路受抑制的情况下,LPS可介导巨噬细胞凋亡;LPS联合三磷酸腺苷(adenosine triphosphate,ATP)可诱导包括巨噬细胞在内的多种细胞发生焦亡[7]。本研究发现,LPS诱导未分化的M0巨噬细胞发生死亡的作用较弱,LDH释放从对照组到LPS(100 μg/L)刺激6 h仅从7.1%上升到9.1%,TUNEL阳性率从(0.3±0.1)%上升到(4.8±0.2)%,这与以往的报道是一致的[8]。然而,当使用IFN-γ分化诱导的M1亚型巨噬细胞在接受LPS刺激6 h后,TUNEL阳性率达峰值(12.9%),而LDH释放在12 h达峰值(22.3%),2种死亡检测指标峰值时间的差异提示LPS诱导M1亚型巨噬细胞发生凋亡,随着时间延长,晚期凋亡细胞的细胞膜出现破坏,导致LDH释放进一步升高。
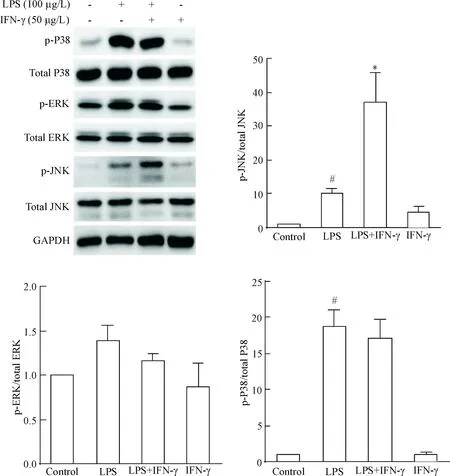
Figure 4. IFN-γ pretreatment enhanced LPS-induced activation of JNK in macrophages. Mean±SD.n=3.#P<0.05vscontrol group;*P<0.05vsLPS group.
图4IFN-γ预处理增强LPS介导的JNK蛋白磷酸化
为进一步明确其死亡方式,本研究使用了多种抑制剂进行了初步验证,对于IFN-γ预处理的M1亚型巨噬细胞,单独caspase-1抑制剂和RIP1抑制剂均不能抑制LPS所介导的LDH释放和DNA断裂。泛caspase抑制剂z-VAD-FMK可阻断LPS介导的DNA断裂,但反而使得其LDH 释放增加,联合使用z-VAD-FMK和necrostatin-1则可完全阻断LDH释放和DNA断裂。因此,LPS介导的M1巨噬细胞死亡考虑为凋亡,LPS联合z-VAD-FMK所介导的M1亚型巨噬细胞考虑为程序性坏死。
程序性坏死是由2个关键分子RIP1和RIP3参与调控的细胞死亡方式,已证明多种刺激信号可以诱导程序性坏死的发生[9]。LPS与TLR4结合后,通过2个接头分子髓样分化因子88(myeloid differen-tiation factor 88,MyD88)和Toll样受体接头分子1(Toll-like receptor adapter molecule 1, TICAM1)激活NF-κB和MAPK通路,TICAM1的活化可募集RIP1和RIP3形成复合体,在凋亡通路被抑制的情况下(例如某些病毒感染或使用z-VAD-FMK),RIP3被磷酸化并进一步聚集,进而磷酸化混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein,MLKL),磷酸化的MLKL转位至细胞膜最终介导程序性坏死[10]。该过程并不依靠LPS诱导巨噬细胞自分泌所产生的TNF-α[11]。而我们结果显示LPS联合z-VAD-FMK仅能诱导M1亚型巨噬细胞发生程序性坏死,这可能是机体自我保护的一种可能机制,以限制本身行使促炎功能的M1亚型巨噬细胞在接受LPS刺激时进一步增强炎症反应,导致免疫过激。进一步探讨LPS联合z-VAD-FMK诱导巨噬细胞发生程序坏死的为何仅仅特异性地发生于IFN-γ诱导分化形成的M1亚型细胞的机制,发现经IFN-γ诱导分化的M1亚型巨噬细胞具有显著高于M0亚型巨噬细胞中RIP3的表达,而RIP1在2种巨噬细胞亚型中并无显著差异。IFN-γ主要通过与其受体结合激活信号转导和转录激活因子1(signal transducer and activator of transcription 1,STAT1)通路,该通路的激活被报道在小鼠中可以上调MLKL的表达[12], 而其是否参与对RIP1和RIP3转录层面的调控迄今未见报道。因此,IFN-γ是通过激活STAT1直接上调RIP3表达,还是通过分泌其它细胞因子间接促进RIP3表达需要进一步研究。另外,除了上游RIP1和RIP3的活化外,程序性坏死的发生也需要JNK参与,JNK的活化也可与RIP3的活化相互促进[13-15],而LPS主要的靶信号通路就是NF-κB和MAPK通路。因此,我们检测了INF-γ是否对LPS介导的JNK磷酸化有影响,发现IFN-γ对JNK的磷酸化并无直接活化或抑制作用,但却能够显著增强LPS介导的JNK磷酸化。进一步使用RIP3和JNK的抑制剂,发现2者均可阻断LPS联合z-VAD-FMK介导的IFNγ预处理的M1亚型巨噬细胞所发生的的LDH释放,提示RIP3和JNK可能参与程序性坏死的发生。Bulosan等[16]发现IFN-γ能够通过抑制巨噬细胞中MAPK磷酸酶(MAPK phosphatase,MKP)家族即MKP-1、 MKP-2和MKP-4的活性,从而增强ERK、JNK和P38的磷酸化。但本研究中,IFNγ对MAPK通路在没有接受LPS刺激的基线并无明显增强作用,而在接受LPS刺激后,仅仅增强JNK的磷酸化,而对ERK和P38并没有作用,因此在不同的巨噬细胞模型中,IFN-γ还可能存在不同的机制对MAPK通路进行调控。
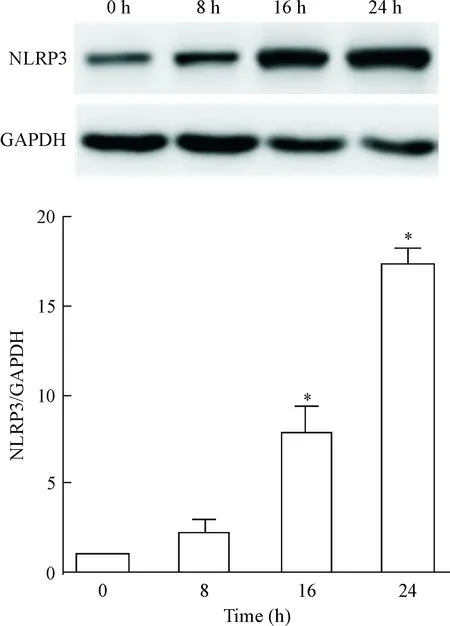
Figure 5. IFN-γ (50 μg/L) upregulated NLRP3 expression in the macrophages. Mean±SD.n=3.*P<0.05vs0 h.
图5IFN-γ上调巨噬细胞NLRP3的表达
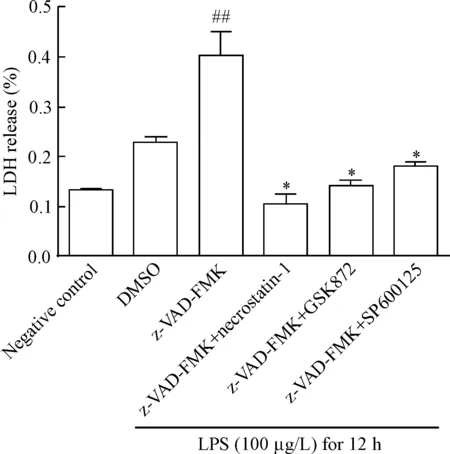
Figure 6. GSK872 (5 μmol/L) and SP600125 (10 μmol/L) significantly blocked the LDH release mediated by LPS combined with z-VAD-FMK in IFN-γ-pretreated macrophages. Mean±SD.n=3.##P<0.01vsDMSO group;*P<0.05vsz-VAD-FMK group.
图6GSK872和SP600125显著阻断LPS联合z-VAD-FMK介导的M1亚型巨噬细胞LDH释放
NLRP3作为被研究的最为广泛的炎症小体,既往的研究主要集中于IL-1β和IL-18的成熟分泌和焦亡中,近期NLRP3还被发现参与JNK的激活,敲除NLRP3会阻断JNK的磷酸化[5],因此我们检测了IFN-γ对NLRP3的影响,发现IFN-γ能够上调巨噬细胞NLRP3的表达,但本研究中NLRP3的上调是否是IFN-γ增强LPS介导的JNK磷酸化的机制有待进一步证实。同时,IFN-γ上调NLRP3的机制也并不清楚,由于IFN-γ激活STAT通路后所涉及调控细胞因子过多,需要在今后的研究中进一步探讨。
综上所述,IFN-γ能够通过增强巨噬细胞RIP3的表达,并可增强LPS介导的JNK磷酸化,使得M1亚型巨噬细胞在LPS联合z-VAD-FMK刺激时发生程序性坏死,从而作为机体一种可能的自我保护机制。
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
皮肤病与性病(2021年3期)2021-07-30
中国动物检疫(2020年1期)2020-01-08
天津医科大学学报(2019年6期)2019-08-13
意林·少年版(2019年11期)2019-06-30
科学之谜(2019年3期)2019-03-28
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
中国当代医药(2015年30期)2015-03-01
