
水中硫代钨酸盐的形成及其形态转化
2018-09-25 01:19郭清海
中国环境科学 2018年9期
赵 倩,郭清海,罗 黎
水中硫代钨酸盐的形成及其形态转化
赵 倩,郭清海*,罗 黎
(中国地质大学环境学院,生物地质与环境地质国家重点实验室,湖北 武汉 430074)
基于紫外-可见分光光度法定量测试了模拟的不同天然水环境条件下硫代钨酸盐的含量变化,探讨了其对硫代钨酸盐的形成和形态转化过程的影响.结果表明:偏酸性-中性条件是水中大量形成一、二、三、四硫代钨酸盐(特别是三、四硫代钨酸盐)的必要前提,且在偏酸性条件下(pH = 5)钨酸盐硫代程度更高.在偏酸性条件下,当硫化物与总钨的摩尔浓度比(S(II)/W)为10:1时,硫代钨酸盐成为钨的主要存在形式;S(II)/W为20:1时,钨酸盐全部转化为硫代钨酸盐,且钨的主要形态是WO3S2-;S(II)/W为30:1和40:1时,钨的主要形态分别是WO2S22-和WOS32-.在偏酸性条件下,离子强度的增加对钨酸盐的硫代过程有明显抑制作用;而在中性条件下,离子强度变化对钨的形态变化的影响甚微.
硫代钨酸盐;形态转化;紫外-可见分光光度法;水环境
钨(W)属于第六周期副族过渡金属元素长期以来被认为是一种无毒、在环境中迁移能力低的惰性金属[1-2],并常作为铅的替代品使用[2-3].虽然到目前为止尚未证实儿童白血病群发与该群体对钨的摄入之间存在确切联系,但实验室动物实验研究确已表明钨有毒、可致癌[4-8],且在中性-偏碱性水环境中为相对保守组分,具有较强的迁移能力[9-10].
自然界中,钨主要以正六价和正四价形式存在,其地球化学行为与其他可形成含氧阴离子的微量元素(如Mo和As)相似,黑钨矿、白钨矿、辉钨矿等为钨的常见矿物.在水环境中,正六价常是溶解态钨的唯一价态.WO42-天然水环境中钨含量通常较低,如海水中钨含量为痕量级,广为接受的海水钨丰度为<0.2~1ng/kg[11-13];河水中钨含量变化范围较大,但未受污染的河水中钨含量一般也小于0.1μg/L[14-15].在某些特殊水环境中,钨可能在水中相对富集,例如海底热液的钨含量与海水相比可提高1~4个数量级.另外,富硫化物天然水中也常常观测到钨含量异常.在美国德克萨斯州的Carrizo含水层中,地下水硫化物浓度达40µmol/L,与马里兰州Aquia含水层中硫化物浓度£2µmol/L的地下水相比,其钨浓度高出16倍以上[16],主要原因是水中高浓度的硫化物使单体钨酸盐WO42-向硫代钨酸盐WOS4-x2-转化,从而增加了钨的溶解度[17].
水中硫代钨酸盐的形成及其对钨的地球化学行为的重要影响与硫代钼酸盐、硫代砷酸盐、硫代锑酸盐等非常相似.例如,文献报道硫代砷酸盐的形成同样可增加水中溶解态砷的总含量[17-21].然而,与钼、砷、锑等其他亲铁元素相比,钨的含氧阴离子的硫代产物(即硫代钨酸盐)的研究程度低得多.因此,在本次研究中,基于紫外-可见(UV/VIS)分光光度法定量检测了不同天然水环境模拟条件(包括不同pH值、硫化物含量、离子强度)下硫代钨酸盐的含量变化,探讨了上述水环境条件对硫代钨酸盐的形成和形态转化过程的影响.
1 材料与方法
1.1 试剂、仪器和分析方法
主要试剂及其来源:Na2WO4·2H2O(分析纯,国药集团化学试剂有限公司);Na2S·9H2O(分析纯,上海统亚化工科技发展有限公司);HCl(优级纯,中国平煤神马集团开封东大化工有限公司);NaCl(分析纯,国药集团化学试剂有限公司).
硫代钨酸盐的定量检测用紫外可见分光光度计(HITACHI U-3900)完成,在手套箱中采集待测样品以尽可能避免S(Ⅱ)氧化,经0.22µm滤头过滤后现取现测,测样的紫外-可见分光光度计配置厚度为1cm的石英比色皿,以超纯水作为空白,测定溶液在270~500nm波长范围内的吸光度.溶液pH值用经标准液校正后的pH计(Mettler Tolerdo FE28)测定,同样在手套箱中现取现测.溶液总钨含量用电感耦合等离子体质谱仪(ICP-MS;7700X型;美国Agilent公司)测定,RF功率1550W,载气流量0.99L/min,检测离子/=184.
1.2 实验过程
为避免Na2S·9H2O中S(Ⅱ)被氧化,实验过程中所有溶液的配制均使用经过高纯氮气(纯度³99.999%)脱氧处理1h以上的超纯水,配制和保存过程均在手套箱中完成.设置3个平行试验.
1.2.1 Na2WO4•2H2O、Na2S•9H2O、NaCl摩尔吸收系数的测定 采用比尔定律计算物质的摩尔吸收系数:以溶液浓度(mol/L)为横坐标,以厚度为1cm比色皿测得的某一波长下吸光度为纵坐标,则该物质在波长下的摩尔吸收系数为拟合直线的斜率.实验配置的Na2WO4·2H2O溶液浓度梯度为1,10,100,500mmol/L;配置的Na2S·9H2O溶液浓度梯度为1,10,100, 500mmol/L;配置的NaCl溶液浓度梯度为10,100, 500,1000mmol/L.
1.2.2 pH值调节剂、盐度调节剂及相关溶液配制
(1)pH值和盐度调节剂:分别选用6.8mmol/L HCl溶液(配制0.1mmol/L Na2WO4·2H2O,S(II)/W = 40:1的混合溶液,调节初始pH=5时所需的HCl,即0.11mL 36%的HCl定容于250mL PET瓶中)和5g/L NaCl溶液作为pH值和盐度调节剂的代表,测定其吸光度.
(2)相关溶液:①将0.025mmol Na2WO4·2H2O和0.11mL 36%HCl定容于250mL PET瓶中;②将1mmol Na2S·9H2O和0.11mL 36%HCl定容于250mL PET瓶中,塞紧瓶口并进行封口处理;③将1.25gNaCl、0.025mmol Na2WO4·2H2O和0.11mL 36%HCl定容于250mL PET瓶中;④将1.25gNaCl、1mmol Na2S·9H2O和0.11mL 36%HCl定容于250mL PET瓶中,塞紧瓶口并进行封口处理.
1.2.3 不同pH值条件下钨的存在形态分析 (1)在装有100mL水的烧杯中溶入1mmol Na2S·9H2O及0.025mmol Na2WO4·2H2O,利用容量瓶将混合液定容至250mL,然后移液至厌氧瓶中.
(2)重复步骤(1)6次,利用36%HCl分别调节溶液初始pH=1、3、5、7、9、11,封口后置于(21±1.5) ℃手套箱中,得到S(II)/W =40:1、无氧条件下,初始pH=1、3、5、7、9、11的钨酸钠与硫化钠混合溶液,反应至预定时间后,测定其一、二、三、四硫代钨酸盐含量,并计算钨酸盐含量.
1.2.4 不同S(II)/W条件下钨的存在形态分析 (1)将0.25,0.5,0.75,1mmol的Na2S·9H2O分别溶于装有100mL水的4个烧杯中,待其完全溶解后分别加入0.025mmol Na2WO4·2H2O,利用容量瓶分别将这4个混合液定容至250mL,移液至4个厌氧瓶中.
(2)利用36%HCl调节四种溶液初始pH=5,封口后置于(21±1.5)℃手套箱中,得到初始pH=5,S(II)/W分别为10:1、20:1、30:1、40:1的钨酸钠与硫化钠混合溶液.
(3)重复步骤(1),然后利用36%HCl调节溶液初始pH=7,封口后置于(21±1.5)℃手套箱中,得到初始pH=7的无氧条件下,S(II)/W分别为10:1、20:1、30:1、40:1的钨酸钠与硫化钠混合溶液.反应至预定时间后,测定其一、二、三、四硫代钨酸盐含量,并计算钨酸盐含量.
1.2.5 不同离子强度条件下钨的存在形态分析 (1)在5个各装有100mL水的烧杯中,分别加入0,0.25,1.25,2.5,12.5g NaCl,然后均加入1mmol Na2S·9H2O和0.025mmol Na2WO4·2H2O,利用容量瓶分别将这5个混合溶液均定容至250mL,移液至5个厌氧瓶中.
(2)利用36%HCl调节三种混合溶液pH=5,封口后置于(21±1.5)℃手套箱中,得到S(II)/W=40:1、初始pH=5的无氧条件下,NaCl=0,1,5,10,50g/L的钨酸钠与硫化钠混合溶液.
(3)重复步骤(1),利用36%HCl调节溶液初始pH=7,封口后置于(21±1.5)℃手套箱中,得到S(II)/W=40:1、初始pH=7的无氧条件下,NaCl浓度分别为0,1,5,10,50g/L的钨酸钠与硫化钠混合溶液,反应至预定时间后,测定其一、二、三、四硫代钨酸盐含量,并计算钨酸盐含量.
1.3 分析方法
本研究基于紫外-可见(UV/VIS)分光光度法计算硫代钨酸盐的浓度,紫外-可见分光光度法在复合材料催化活性、溶解性有机质、污泥中辅酶等定量检测中应用广泛[22-24].据朗伯-比尔定律,混合溶液产生的吸光度具有可加性.因此,在给定波长条件下,本研究中基于紫外-可见(UV/VIS)分光光度法测得的吸光度是溶液中各种硫代钨酸盐的浓度的函数,可表示为:
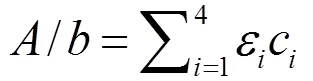
式中:为吸光度;为吸收层厚度(cm);ε和c(= 1、2、3、4)分别为一、二、三、四硫代钨酸盐的摩尔吸收系数及其在待测溶液中的浓度.
获得待测溶液在270~500nm波长范围的吸光度后(硫代钨酸盐溶液的吸光度测试曲线示例见附图1),利用4种硫代钨酸盐在该波长范围内的摩尔吸收系数[20],可通过经典最小二乘法和约束最小二乘法[25]计算溶液中4种硫代钨酸盐的浓度.溶液中WO42-浓度用差减法计算,即钨酸盐含量为总钨含量减去4种硫代钨酸盐含量.当用经典最小二乘法计算得到的4种硫代钨酸盐的浓度总和大于总钨含量时,可认为溶液中钨酸盐已全部转化为硫代钨酸盐,此时可利用约束最小二乘法计算溶液中4种硫代钨酸盐的浓度.
2 结果与讨论
在本研究中,为探讨硫代钨酸盐在不同环境条件下的形成和形态转化规律,需调节实验室配制溶液的pH值、离子强度等水化学参数,因此需在溶液中加入pH值调节剂(使用HCl)和离子强度调节剂(使用NaCl).此外,用于形成硫代钨酸盐的Na2WO4和Na2S在大多数情况下不可能完全反应,在测试吸光度时仍残留在溶液中,故同样有必要评价Na2WO4和Na2S对硫代钨酸盐的吸光度测试的影响.
2.1 溶液中相关组分的摩尔吸收系数及其在给定浓度下产生的吸光度
Na2WO4、Na2S、NaCl和4种硫代钨酸盐在270~ 500nm波长范围内的摩尔吸收系数(L/(mol·cm))见图1(a).与一、二、三、四硫代钨酸盐相比,Na2S溶液的摩尔吸收系数在该波长范围内非常低,而NaCl、Na2WO4溶液的摩尔吸收系数则为0.图1(b)为HCl、NaCl、Na2WO4、Na2S溶液在270 ~ 500nm波长范围内产生的吸光度,上述试剂除了Na2S在给定波长范围内产生了较低的吸光度之外,其它试剂几乎不产生吸光度.

2.2 pH值和盐度调节剂与反应物(Na2WO4、Na2S)的混合溶液产生的吸光度
图2(a)显示,Na2WO4溶液和HCl在270 ~ 500nm波长范围内产生的吸光度为0,但二者的混合溶液在相同波长范围内产生了吸光度,因此它们之间应发生了某种反应,即HCl的加入可能干扰钨酸盐的硫代过程.
据图2(b),HCl与Na2S溶液混合后所得吸光度曲线与Na2S溶液吸光度曲线相似,由于HCl在270~500nm波长范围内不产生吸光度,HCl的加入应不明显影响溶液中Na2S参与钨酸盐的硫代反应,也不会对硫代钨酸盐定量检测产生干扰.

图2 各组分的不同组合的吸光度在270~500nm波长范围内的变化
A(X)+A(Y)代表溶液X和溶液Y各自产生的吸光度之和,A(X+Y)代表X、Y混合溶液产生的吸光度
图2(c)显示NaCl-Na2WO4-HCl 3者混合溶液产生的吸光度与Na2WO4-HCl混合液产生的吸光度变化趋势一致,但有微弱减小趋势,说明NaCl对Na2WO4-HCl混合液产生的吸光度有影响,但并不是NaCl参与Na2WO4-HCl混合溶液反应造成的,而是由于NaCl的加入使得Na2WO4-HCl的有效离子碰撞数量减小,抑制了二者之间的反应,导致Na2WO4-HCl混合液产生的吸光度略有降低.
由图2(d)可知,Na2S-HCl混合溶液与NaCl溶液产生的吸光度之和与Na2S溶液的吸光度接近,而三者混合后吸光度显著降低.原因是:离子强度增加(加入NaCl所致)改变了S2-水解反应的平衡[26],水解生成的在选定波长范围内具有吸光度的HS-和H2S[27-28]浓度降低,从而导致混合后溶液的吸光度值降低.NaCl的加入有利于消除S2-水解生成的HS-和H2S产生的吸光度对硫代钨酸盐吸光度测试的干扰.
HCl因与Na2WO4发生某种反应可能会干扰钨酸盐的硫代过程,而对Na2S几乎没有影响;NaCl可使Na2WO4-HCl混合溶液的吸光度略有降低,但影响极小,而使Na2S-HCl混合溶液的吸光度明显降低,这有利于硫代钨酸盐吸光度的测试,但因改变了S2-的水解过程而对钨酸盐的硫代过程可能会有影响.综上,Na2S、HCl、NaCl会对钨酸盐的硫代反应以及硫代钨酸盐的吸光度测试产生影响.
根据朗伯-比尔定律计算硫代钨酸盐浓度时,采用数学统计方法中的拟合优度检验系数即拟合系数2来表征计算结果的可靠程度.2是指利用朗伯-比尔定律算出的各物质浓度与对应摩尔吸收系数的乘积(拟合吸光度)之和与测得的溶液吸光度之间的拟合系数,其计算公式如下:
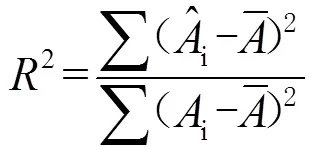
2.3 pH值对水中钨的存在形态的影响
pH值是影响天然水中硫代化合物形态分布的重要因素.例如,已有研究表明硫代砷酸盐在中性-偏碱性的富硫化物天然水中是砷的主要存在形态,而在酸性含硫化物天然水中则含量很低,仅为次要存在形态[29-30].为了比较硫代钨酸盐与研究程度较高的硫代砷酸盐的形成过程对环境pH值的响应差异,首先分析了pH值对水中钨的形态分布的控制作用.

偏酸性-中性条件是水中大量形成一、二、三、四硫代钨酸盐(特别是三、四硫代钨酸盐)的必要前提.在pH£3时,一硫代钨酸盐是硫代钨酸盐的主要存在形式;在偏碱性条件下,即便溶液中S(II)/W高达40:1,钨酸盐仍是钨的主要存在形式,水中虽可检出一硫代钨酸盐,但二、三、四硫代钨酸盐含量极低.因此,硫代钨酸盐的存在形态对水环境pH值的响应明显有异于研究程度较高的硫代砷酸盐.Planer- Friedrich等[29]在美国Yellowstone地热区的研究中发现在中性-偏碱性热泉中硫代砷酸盐可占到总砷含量的83%,而酸性热泉中硫代砷酸盐在总砷中的百分含量则低得多.Planer-Friedrich[31]基于室内实验研究指出四硫代砷酸盐只存在于强碱性环境中,在pH<11时就会完全转化为三硫代砷酸盐.pH值对水中硫代钨酸盐的影响在本质上是水中羟基和巯基在钨酸盐结构中的竞争关系的反映.本文中虽为方便起见,将各种硫代钨酸盐的化学结构式写做其脱质子形式,但事实上每一种硫代钨酸盐均存在多种质子化形式,如写做WO3S2-的一硫代钨酸盐应包括WO3S2-、HWO3S-(即WO3(SH)-)、H2WO3S(即WO2(OH)(SH))三种形式.这样,与碱性条件相比,酸性条件下水中SH-相对于OH-的优势更加明显,钨酸盐的硫代过程(即其结构中OH-被SH-取代的过程)也更易发生.
2.4 水中硫化物的存在对钨的形态分布的影响
水中硫化物含量是砷、钼、钨等亲铁元素的形态分布的控制因素之一[17,32-33].研究表明:硫化物的富集是天然水中硫代钼酸盐[34]和硫代砷酸盐(特别是高硫代砷酸盐)[29-30]大量存在的必要条件;硫化物驱动下水中钨酸盐的硫代过程也控制着硫代钨酸盐的形成,如以下反应式[17]所示:
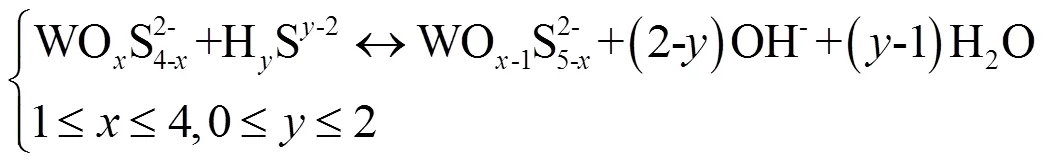
上述反应方程指示硫化物浓度的增加将促进钨酸盐的硫代反应.
地球化学模拟显示,与硫代钼酸盐相比,硫代钨酸盐的形成需要大约40倍浓度的硫化物[17].尽管如此,某些富硫化物-强还原性天然水(如静海相沉积物孔隙水、岩浆热源型地热水)中仍可能存在足够的硫化物以形成钨酸盐的硫代产物.鉴于国内外均缺乏天然水中硫代钨酸盐的定量检测方法,当前罕有富硫化物天然水中硫代钨酸盐存在的实测证据.因此,在此探讨了实验室控制条件下水中硫化物-总钨的摩尔浓度比(S(II)/W)对钨的形态分布的实际影响.


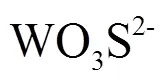
2.5 离子强度对水中钨存在形态的影响
富硫化物天然水的总溶解固体(TDS)具有宽广的范围.例如,西藏山南地区曲松县竹墨沙热泉(硫化物含量:1.36mg/L)的TDS为9.05g/L,昌都地区察雅县达坝热泉(硫化物含量:37.62mg/L)的TDS为3.18g/L,而西藏昌都地区丁青县琼卡热泉(硫化物含量:7.75mg/L)的TDS仅为0.32g/L.水中离子强度的差异可能也会影响硫代钨酸盐的形态分布.因此,本次研究配制了不同含盐量的溶液(NaCl浓度分别为0,1,5,10,50g/L),其中离子强度是根据所配置溶液的实际水化学参数利用Phreeqc软件计算得出,以评价离子强度对水中钨的形态分布的可能影响.


图5 不同离子强度下酸性水溶液中钨的形态分布随反应时间的变化
综上,离子强度的增加在酸性条件下(溶液初始pH=5)对钨酸盐的硫代过程有明显抑制作用,当离子强度为25mmol/L时硫代钨酸盐占总钨的比例就明显下降,且硫代钨酸盐的硫代程度相应降低;而在中性条件下(溶液初始pH=7),离子强度变化对钨的形态分布的影响不明显.
3 结论
3.1 偏酸性-中性条件是水中大量形成硫代钨酸盐(特别是三、四硫代钨酸盐)的必要前提,且硫代钨酸盐占总钨的比例及硫代钨酸盐的硫代程度随水中S(II)/W的增加而增加.
3.2 在偏碱性条件下,即便溶液中S(II)/W高达40:1,钨酸盐仍是钨的主要存在形式,水中虽可检出一硫代钨酸盐,但二、三、四硫代钨酸盐含量极低.
3.3 离子强度的增加在酸性条件下会抑制硫代钨酸盐的形成,但在中性条件下无明显影响.
[1] Koutsospyros A, Braida W, Christodoulatos C, et al. A review of tungsten: from environmental obscurity to scrutiny [J]. Journal of Hazardous Materials, 2006,136(1):1-9.
[2] Bednar A J, Mirecki J E, Inouye L S, et al. The determination of tungsten, molybdenum, and phosphorus oxyanions by high performance liquid chromatography inductively coupled plasma mass spectrometry [J]. Talanta, 2007,72(5):1828-1832.
[3] Strigul N, Koutsospyros A, Arienti P, et al. Effects of tungsten on environmental systems [J]. Chemosphere, 2005,61(2):248-258.
[4] Sheppard P R, Ridenour G, Speakman R J, et al. Elevated tungsten and cobalt in airborne particulates in Fallon, Nevada: Possible implications for the childhood leukemia cluster [J]. Applied Geochemistry, 2006,21(1):152-165.
[5] Sheppard P R, Speakman R J, Ridenour G, et al. Temporal variability of tungsten and cobalt in Fallon, Nevada [J]. Environmental Health Perspectives, 2007,115(5):715–719.
[6] Strigul N. Does speciation matter for tungsten ecotoxicology? [J]. Ecotoxicology and Environment Safety, 2010,73(6):1099-1113.
[7] Strigul N, Koutsispyros A, Christodoulatos C. Tungsten speciation and toxicity: acute toxicity of mono- and poly-tungstates to fish [J]. Ecotoxicology and Environment Safety, 2010,73(2):164–171.
[8] Kelly A D R, Lemaire M, Young Y K, et al. In vivo tungsten exposure alters B-cell development and increases DNA damage in murine bone marrow [J]. Toxicological Sciences, 2013,131(2):434-446.
[9] Clausen J L and Korte N. Environmental fate of tungsten from military use [J]. The Science of the Total Environment, 2009, 407(8): 2887-2893.
[10] Johannesson K H, Tang J. Conservative behavior of arsenic and other oxyanionforming Trace elements in an oxic groundwater flow system [J]. Journal of Hydrology, 2009,378(1/2):13-28.
[11] 马东升.钨的地球化学研究进展 [J]. 高校地质学报, 2009,15(1): 19-34.
[12] Turekian K K. Oceans [M]. Englewood Cliffs, New York: Prentice Hall, 1968:120.
[13] Kunzendorf H, Glasby G P. Tungsten accumulation in Pacific ferromanganese deposits [J]. Mineralium Deposita, 1992,27(2): 147-152.
[14] Krauskopf K B. Tungsten. In: Wedepohl K H (Ed.). Handbook of Geochemistry [M]. New York: Springer, 1970,2(5):Chapter 74B-O.
[15] Van der Sloot H, Hoede D, Wijkstra J, et al. Anionic species of V, As, Se, Mo, Sb, Te and W in the Scheldt and Rhine estuaries and the Southern Bight (North Sea) [J]. Estuarine, Coastal and Shelf Science, 1985,21(5):633-651.
[16] Johannesson K H, Dave H B, Mohajerin T J, et al. Controls on tungsten concentrations in groundwater flow systems: the role of adsorption and thiotungstate formation [J]. Chemical Geology, 2013,351:76-94.
[17] Mohajerin T J, Helz G R, White C D, et al. Tungsten speciation in sulfidic waters: determination of thiotungstate formation constants and Determination of thiotungstate formation constants and modeling their distribution in natural waters [J]. Geochimica et Cosmochimica Acta, 2014,144:157-172.
[18] Lee M K, Saunders J A, Wilkin R T, et al. Geochemical modeling of arsenic speciation and mobilization: implications for bioremediation [J]. Acs Symposium Series, 2009,915:398-413.
[19] Couture R M, Rose J, Kumar N, et al. Sorption of arsenite, arsenate and thioarsenates to iron oxides and iron sulfides: a kinetic and spectroscopic investigation [J]. Environmental Science & Technology, 2013,47(11):5652-5659.
[20] Mamindy-Pajany Y, Bataillard P, Seby F, et al. Arsenic in marina sediments from the Mediterranean Coast: speciation in the solid phase and occurrence of thioarsenates [J]. Soil and Sediment Contamination, 2013,22(8):984-1002.
[21] Yang N, Welch K A, Mohajerin T J, et al. Comparison of arsenic and molybdenum geochemistry in meromictic lakes of the McMurdo Dry Valleys, Antarctica: implications for oxyanion-forming trace element behavior in permanently stratified lakes [J]. Chemical Geology, 2015,404:110-125.
[22] 张钱新,王枫亮,谢治杰,等.摻N碳量子点负载于TiO2的复合催化剂光解甲芬那酸研究 [J]. 中国环境科学, 2017,37(8):2930-2940.
[23] 吴唯民,胡纪萃,顾夏声.厌氧污泥中的辅酶F420及其紫外-可见光分光光度法测定 [J]. 中国环境科学, 1986,6(1):65-69.
[24] 闫金龙,江 韬,赵秀兰,等.含生物质炭城市污泥堆肥中溶解性有机质的光谱特征 [J]. 中国环境科学, 2014,34(2):459-465.
[25] Montgomery D C, Peck E A, Vining G G. Introduction to linear regression analysis [M]. In Wiley Series in Probability and Statistics: Wiley, 2012, p672.
[26] Hershey J R, Plese T, Millero F J. The pK sub 1* for the dissociation of H2S in various media [J]. Geochimica et Cosmochimica Acta, 1987,52(8):2047-2051.
[27] Linkous C A, Huang C, Fowler J R. UV photochemical oxidation of aqueous sodium sulfide to produce hydrogen and sulfur [J]. Journal of Photochemistry and Photobiology A: Chemistry, 2004,168(3): 153-160.
[28] Zhang X X, Cui Z L, Cheng Z, et al. Quantitative detection of H2S and CS2mixed gases based of UV absorption spectrometry [J]. Poyal Society of Chemistry, 2017,7(80):50889-50898.
[29] Friedrich B P, London J, Mccleskey R B, et al. Thioarsenates in Geothermal Waters of Yellowstone National Park: Determination, Preservation, and Geochemical Importance [J]. Environmental Science & Technology, 2007,41(15):5245-5251.
[30] Guo Q H, Planer-Friedrich B, Liu M L, et al. Arsenic and thioarsenic species in the hot springs of the Rehai magmatic geothermal system, Tengchong volcanic region, China. Chemical Geology, 2017,453: 12-20.
[31] Friedrich B P, Wallschläger D. A critical investigation of hydride generation-based arsenic speciation in sulfidic waters [J]. Environmental Science and Technology, 2009,43(13):5007-5013.
[32] Erickson B, Helz G R. Molybdenum (VI) speciation in sulfidic waters: stability and lability of thiomolybdates [J]. Geochimica et Cosmochimica Acta, 2000,64(7):1149-1158.
[33] Stauder S, Raue B, Sacher F. Thioarsenates in sulfidic waters. Environmental science & technology, 2005,39(16):5933-5939.
[34] Lohmayer R, Reithmater G M S, Bura-Nakić E, et al. Ion-Pair Chromatography Coupled to Inductively Coupled Plasma-Mass Spectrometry (IPC-ICP-MS) as a Method for Thiomolybdate Speciation in Natural Waters. Analytical chemistry [J]. 2015,87(6): 3388-3395.
Formation and species transformation of aqueous thiotungstates.
ZHAO Qian, GUO Qing-hai*, LUO Li
(State Key Laboratory of Biogeology and Environment Geology, School of Environmental Studies, China University of Geosciences, Wuhan 430074, China)., 2018,38(9):3437~3446
The concentrations of thiotungstates formed under various experimental conditions simulating natural aquatic environments were quantitatively determined by an ultraviolet/visible spectrophotometer, with their effects on the formation and species transformation of thiotungstates being evaluated. The results demonstrate that weakly acidic to neutral pH values were necessary for the substantial formation of mono-, di-, tri-, and tetra-thiotungstates, especially tri-and tetra-thiotungstates, and the thiolation of tungstate to the highest degree occurred at pH = 5. Under acidic conditions, thiotungstates were prone to be the major species of tungsten even at relatively low molar ratios of sulfide to tungsten (e.g. S(II)/W ratio = 10:1).With an increase of S(II)/W ratio to 20:1, complete thiolation of tungstate was observed, and the dominant species of tungsten became mono-thiotungstate WO3S2-. At S(II)/W ratios of 30:1and 40:1, di-and tri-thiotungstates WOS32-predominated in the solutions, respectively. Moreover, for the acidic solutions, the increase of ionic strength obviously inhibited the thiotungstate-forming reactions. In contrast, the variation of solution ionic strength had little effect on the tungsten speciation under neutral conditions.
thiotungstate;species transformation;UV/VIS spectrophotometry;aquatic environment
X502
A
1000-6923(2018)09-3437-10
赵 倩(1993-),女,湖北襄阳人,中国地质大学(武汉)硕士研究生,主要从事环境地球化学领域的研究.
2018-01-25
国家自然科学基金资助项目(41772370);国家自然科学基金资助项目(41572335);中央高校基本科研业务费专项资金资助项目(CUGQYZX1717)
* 责任作者, 教授, qhguo2006@gmail.com
猜你喜欢
电子测试(2022年16期)2022-10-17
当代水产(2021年3期)2021-07-20
上海金属(2021年3期)2021-06-10
地球科学与环境学报(2020年2期)2020-03-26
汽车电器(2019年1期)2019-03-21
陶瓷学报(2019年5期)2019-01-12
天然产物研究与开发(2018年5期)2018-06-13
中国有色金属学报(2018年2期)2018-03-26
分析化学(2016年11期)2017-04-25
中南大学学报(自然科学版)(2016年2期)2017-01-19
