
不同产地连翘药材指纹图谱的研究
2018-09-20 08:59王德民冯甜甜
山东化工 2018年15期
李 敏,王德民,冯甜甜,李 峰,冯 帅
(山东中医药大学,山东 济南 250355)
中药指纹图谱是一种综合的,可量化的鉴定手段,它是建立在中药化学成分系统研究的基础上,主要用于评价中药材以及中药制剂半成品质量的真实性、优良性和稳定性的一种方法[1]。高效液相色谱法是目前中药材质量检测应用最广泛的方法,该方法具有分离效率高、分析速度快、精密度高、检测器类型多、方法稳定性好等诸多特点,而且对样品无挥发性和热稳定性方面的限制,比气相色谱法应用要广泛的多,中药材中绝大多数的化学成分均可釆用该方法进行分析,是目前构建中药指纹图谱的主要方法[2]。目前,市售连翘药材的产地加工方法有生晒、蒸、煮三种,不同的产地与加工方法使得连翘药材质量差异较大。本研究目的是为连翘药材质量的评价提供实验依据,故选择了药材含量丰富的50%甲醇提取液,对连翘药材进行了HPLC指纹图谱分析。
1 试剂、仪器与样品
1.1 试剂 、仪器
高效液相色谱仪(检测器型号:UV-L2400,日立公司);色谱柱(VP-ODS柱,粒度5μm,150 mm×4.6 mm,美国安捷伦科技公司);乙醇(分析纯,天津市富宇精细化工有限公司,批号:20160924);甲醇(色谱纯,天津市四友卓越科技有限公司,批号:Q/BSYZ01-2016);磷酸(分析纯,天津科工工贸有限公司,批号:150821);乙腈(色谱纯,美国TEDIA);纯净水(娃哈哈公司);连翘苷标准品(中国食品药品检定研究院,批号:110821-201112,纯度96.8%);连翘脂苷A标准品(中国食品药品检定研究院,批号:111810-201112,纯度92.9%);芦丁标准品(中国药品生物制品检定所,批号:111810-201109,纯度90.5%)[2]。
1.2 供试药材

表1 采集供试药材列表

表2 市售供试药材列表
采集12份连翘药材,购买的6批市售药材均经山东中医药大学中药鉴定学教研室李峰教授鉴定,符合《中国药典》2015年版一部规定,为正品青翘[3]。见表1、2[4]。参考《中华人民共和国药典》药材炮制通则中蒸法、煮法项下炮制。参考郑伶俐的《陕西道地药材连翘炮制工艺及质量标准研究》中正交实验法优选出的连翘产地炮制最佳工艺进行实验药材的炮制[5]。
2 方法与结果
2.1 供试品提取溶剂的优选
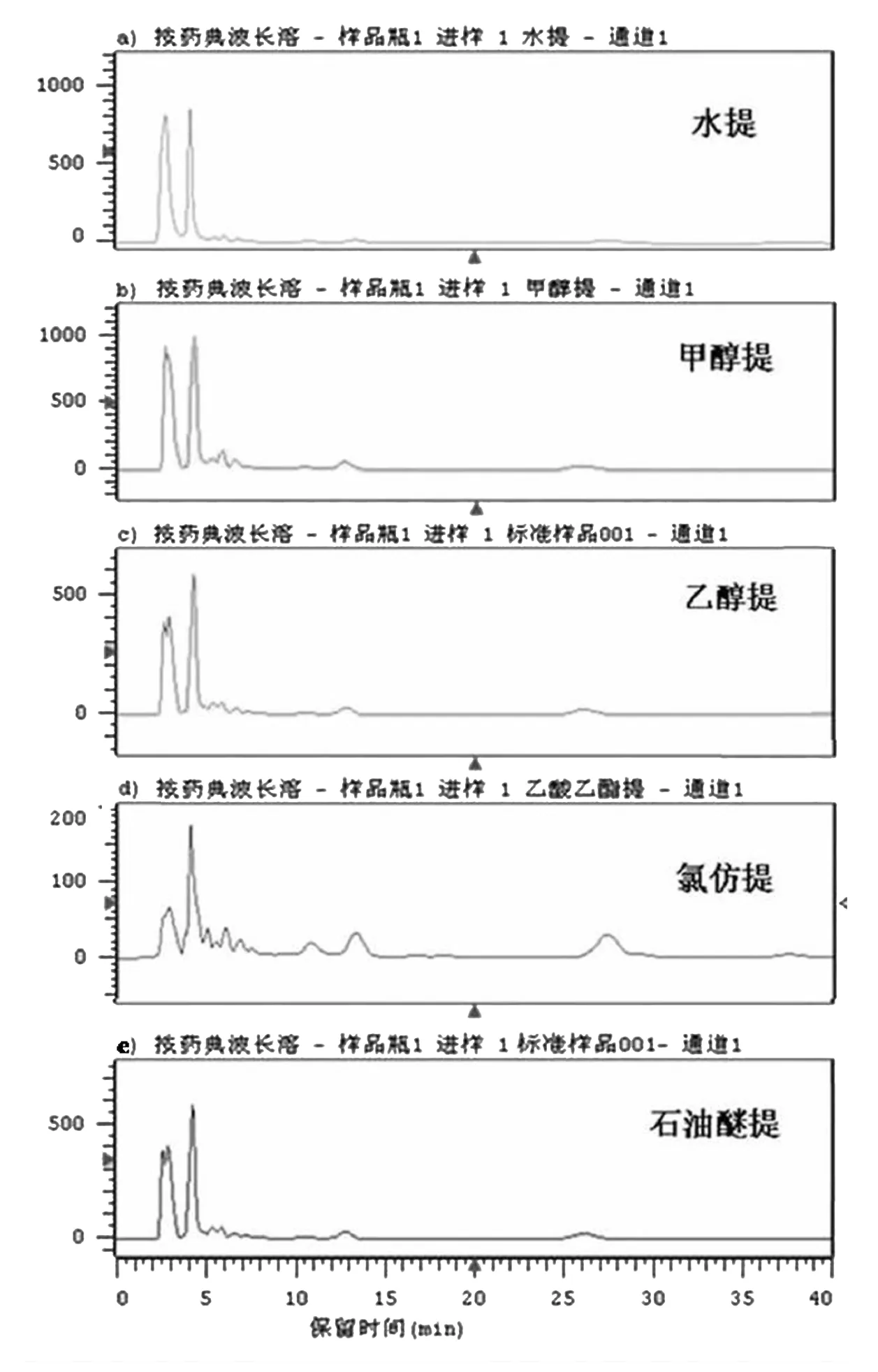
图1 不同提取溶剂提取液的HPLC色谱图
连翘药材经40目筛粉碎成粗粉,精密称取药材粉末0.5 g,分别用15 mL水、乙醇、甲醇、乙酸乙酯、石油醚为提取溶剂,超声振荡提取30min,过滤,后利用旋蒸挥干溶剂,以甲醇定容至10 mL。用0.45 μm微孔滤膜滤过后进行HPLC分析,条件:流速1 mL/min,吸收波长270 nm,记录60min的色谱图,见图1。结果显示甲醇当提取溶剂提取的色谱峰数目较多,峰强度较高。所以进一步将25%、50%、75%、100%甲醇作为提取溶剂,筛选得出50%甲醇为溶剂时各峰信号值最高,见图2。因此选择50%的甲醇为最终的提取溶剂[6]。
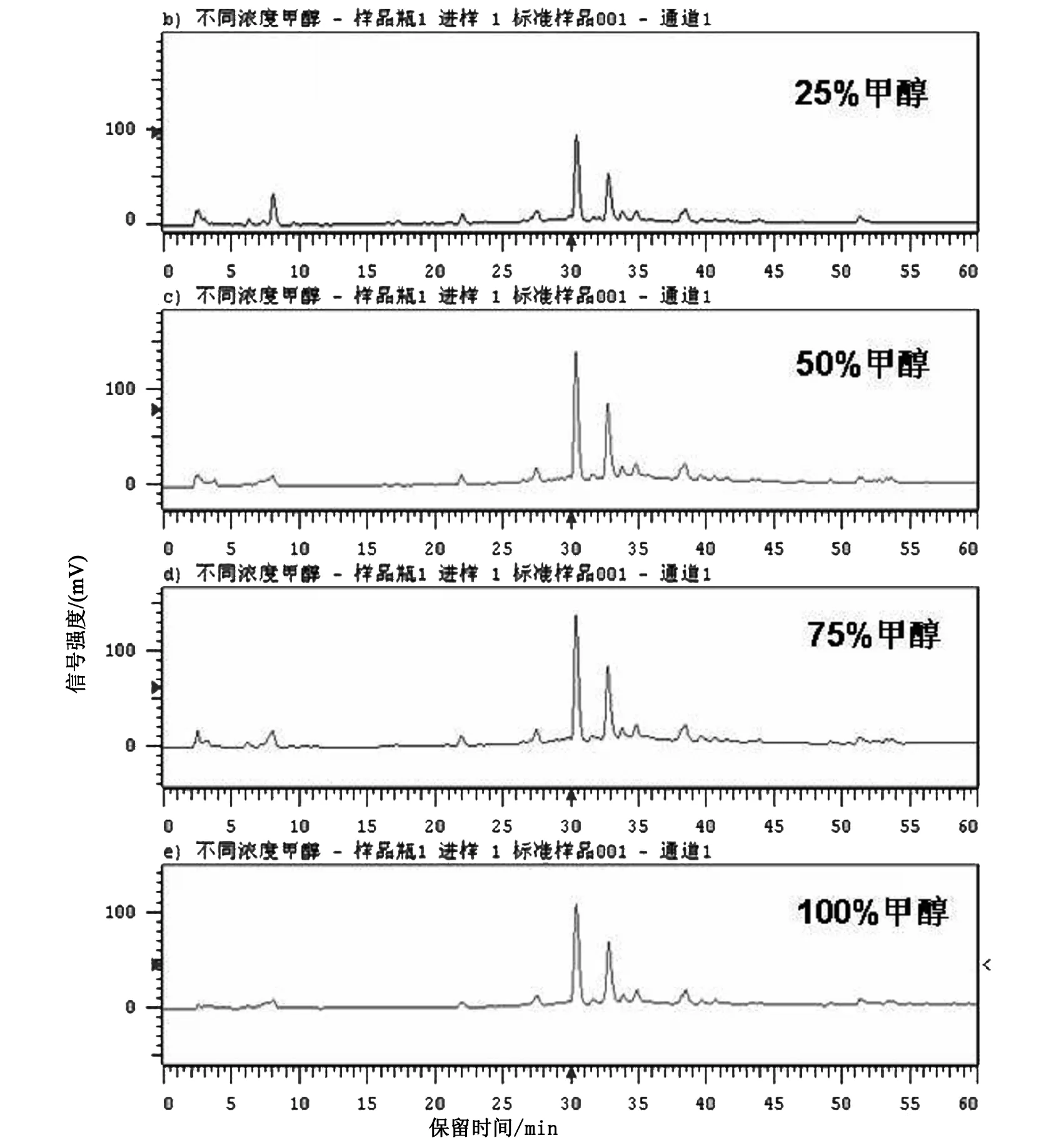
图2 不同浓度甲醇提取液的HPLC图谱比较
2.2 供试品提取时间的选择

图3 不同提取时间的提取液HPLC色谱图比较
精密称取连翘药材粉末0.5g,加入15mL50%甲醇,超声震荡提取。提取时间分别为20、30、40、50min,依上述条件进行液相测定,记录60min的色谱图,筛选得出超声提取30min时各峰信号值最高,见图3。所以最终以超声30min为提取时间。
2.3 高效液相色谱条件的选择
2.3.1 检测波长的选择
以上述优选出的提取溶剂制备样品液,采用乙腈和水梯度洗脱,进行190~400 nm全波长扫描检测[5]。扫描结果见图4。参照全波长扫描结果,选择能够较多的显示连翘化学成分信息的几个检测波长,进一步进行波长优选。如图5,依次为:219、235、270、290 nm。后对上述四个波长进行进一步的优选,根据结果,选择检测波长为235 nm。

图4 连翘50%甲醇提取液的HPLC3D光谱图

图5 检测波长的选择
2.3.2 流动相的选择
精密称取0.5 g药材粉末,加入15mL50%甲醇,超声提取30min,分别以甲醇-水、乙腈-水、乙腈-0.1%磷酸水为流动相,柱温25℃,流速1 mL/min,进样量20μL,检测波长235 nm,记录60min色谱图,结果如图6所示。可见,以乙腈为流动相的色谱峰数较甲醇的色谱峰多,以0.1%磷酸水为流动相的色谱峰分离度较好,因此将流动相定为乙腈-0.1%磷酸水[6]。

图6 不同流动相的HPLC色谱图比较
2.3.3 柱温的选择

图7 不同温度下的HPLC色谱图比较
以上述优选条件进行试验,后分别取柱温20、25、30、35℃进行液相测定,HPLC条件为流速1 mL/min,进样量20μL,检测波长235 nm,记录60min所得色谱图,色谱图如图7所示。可见,当柱温上升到30℃时色谱峰的分离度最好,因此最终将柱温定为30℃。
2.4 连翘HPLC色谱条件的确定
连翘药材粉碎过40目筛,精密称取药材粉末0.5 g置于锥形瓶中,以50%甲醇15 mL超声提取30min,0.45μm微孔滤膜过滤,以甲醇-乙腈-0.1%磷酸水为流动相,梯度洗脱条件见表3,柱温30℃,流速1 mL/min,进样量20μL,检测波长235 nm。色谱图见图8、9。

表3 梯度洗脱条件
2.5 对照品实验
取适量的连翘苷、连翘酯苷A、芦丁的标准品,以50%甲醇溶解,取20μL,按照优选的HPLC色谱条件洗脱,确定对照品的位置,记录色谱图,见图8[7]。

图8 对照品与连翘HPLC色谱图的比对
2.6 方法学考察
同一样品在同一色谱条件下的相对保留时间(RRT)、相对积分面积比值(RA)相对固定,是中药HPLC指纹图谱的两个核心参数,可以作为指纹图谱评价的重要变量。以出峰时间、峰面积较为稳定,且通过标准品对照确定为连翘苷的色谱峰作为参照峰,进行其他色谱峰RRT、RA的计算[8]。
2.6.1 精密度试验
精密称取样品粉末0.5 g,按上述色谱条件,连续进样6次,测定峰面积占总峰面积2%以上的色谱峰的相对保留时间及峰面积。结果表明,峰面积占总峰面积1%以上的色谱峰25个,其相对保留时间的RSD值为0.0104%~0.0461%,峰面积的RSD值为0.9622%~2.6353%,均在3%以内,符合指纹图谱技术要求[9]。
2.6.2 稳定性试验
精密称取样品粉末0.5g,按上述色谱条件,分别在0,2,4,6,8,10 h进样。结果表明,峰面积占总峰面积1%以上的色谱峰25个,其相对保留时间的RSD值为0.0106%~0.3718%,峰面积的RSD值为0.6085%~2.5809%,均在3%以内,符合指纹图谱技术要求。
2.6.3 重现性试验
精密称取样品粉末0.5 g共6份,分别按上述色谱条件测定。结果表明,峰面积占总峰面积1%以上的色谱峰25个,其相对保留时间的RSD值为0.0136%~0.9122%,峰面积的RSD值为0.7097%~2.9175%,均在3%以内,符合指纹图谱技术要求[10]。
2.7 结果
2.7.1 12批不同产地连翘药材的检测
按2.4法,对12批不同产地的连翘药材(经蒸、煮、生晒加工共22个样)进行HPLC检测,利用《中药色谱指纹图谱相似度评价系统2004A版》[11]对所得HPLC图谱进行了共有峰的比对和标定,20个共有峰,见图9、10,共有峰保留时间见表4。所有样品的共有峰面积和均占总峰面积的90%以上。
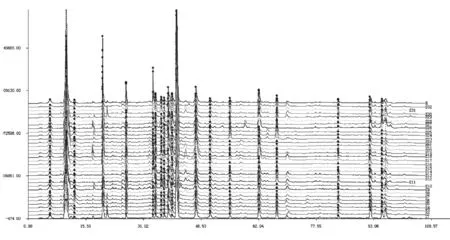
图9 不同产地不同加工方式连翘药材的HPLC色谱图比对

图10 连翘HPLC对照谱图

编号保留时间/min相对保留时间编号保留时间/min相对保留时间15.8830.0951138.7880.624210.2400.1651239.8870.642312.4810.2011345.1360.727420.0240.3221448.9570.788526.3310.4241554.3000.874633.6090.5411662.1151.000734.3000.5521766.8891.077835.8260.5771883.4691.344936.6330.5901991.9681.4811037.7030.6072095.2611.534
2.7.2 6批市售连翘的检测
按照前述的HPLC色谱条件,对6批市售连翘药材进行检测,绘制的HPLC如图11。

图11 不同产地市售连翘药材的HPLC色谱图
利用《中药色谱指纹图谱相似度评价系统2004A版》,以前期试验所得的HPLC对照指纹图谱谱图为参照,对所得HPLC图谱进行了共有峰的比对和标定,结果见图12[12]。6批药材所得的HPLC色谱图与前期得到的连翘药材HPLC对照指纹图谱的相似度评价,见表5。结果显示,6批市售药材与已知HPLC对照指纹图谱的相似度均>0.90,可见前期试验所得的HPLC对照指纹图谱对于连翘药材的鉴别具有很好的应用性。
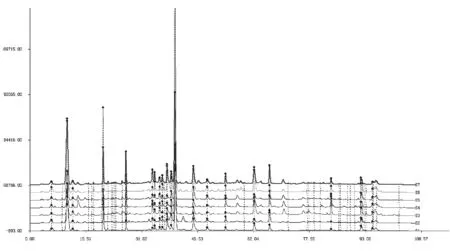

1 HPLC对照指纹图谱,2 河北,3河南,4 济南,5 山西,6 陕西,7 泰山
3 小结与讨论
3.1 连翘HPLC色谱条件的选择
3.1.1 提取溶剂的选择
本次实验通过选用不同的提取溶剂,即水、甲醇(25%、50%、75%、100%)、乙腈、乙酸乙酯、石油醚等,从而筛选得出以50%的甲醇做提取液时所得到的色谱峰数目较多,而且各峰信号值也较高。所以,最终确定以50%甲醇为提取溶剂,结果显示连翘样品50%甲醇提取液能够较好的反应其整体的化学成分,也能够较全面地反映出其所含化学成分浓度分布的状况。
3.1.2 洗脱溶剂的选择
连翘的50%甲醇提取液的成分比较复杂,所以单一的使用一种流动相很难实现很好的分离效果,因此本实验采用了梯度洗脱的方式。通过初步研究,得出乙腈作为流动相的色谱峰大于甲醇的色谱峰,以0.1%磷酸作为流动相分离色谱峰优于单乙腈。在分析过程中,发现在运行时间为10min时,色谱峰不完全分离,如果5%乙腈洗脱,则会出现峰积累现象。所以这一部份应选择洗脱性比乙腈弱的甲醇为流动相。从而使样品能够得到良好的分。经过多次反复实验,最终确用流动相为乙腈-0.1%磷酸水进行梯度洗脱[15 ]。
3.1.3 检测波长的选择
在本实验中,比较了不同波长检测的HPLC谱峰的色谱峰数、分离度和吸收强度。结果,随着检测波长的提高,连翘提取液的HPLC色谱峰信息会越来越稀少。经过进一步比较优化,筛选出最佳检测波长,即以235 nm作为检测波长[16]。
3.1.4 洗脱温度的选择
本次实验以5℃为梯度分别取柱温20~30℃进行了HPLC检测,结果表明,当柱温为30℃时,各色谱峰的分离效果较好。但柱温升高至35℃时,色谱峰出现堆叠,分离效果变差。经综合分析后,最终确定柱温为30℃。综上,经反复比较优选,最终确定了连翘指纹图谱的研究条件。
3.2 讨论
3.2.1 不同产地加工方式连翘的HPLC图谱分析
本研究对经三种产地加工处理的三批连翘药材生晒品、蒸品、煮品进行了HPLC检测,比对结果显示三种产地加工品的HPLC图谱相似性较高;显示蒸、煮加工虽然会导致有些化学成分的损失、降解或性质的改变,但主化学成分未发生变化[17]。因此,本研究确定的连翘HPLC指纹图谱对连翘药材具有很好的适用性、推广性。
3.2.2 连翘HPLC指纹图谱的适用性研究
利用《中药色谱指纹图谱相似度评价系统2004A版》,以研究建立的连翘HPLC指纹图谱方法,对6批市售连翘药材所得的HPLC色谱图进行了鉴别和相似度评价[18]。结果显示,6批市售药材与已知HPLC对照指纹图谱的相似度均>0.90,可见前期试验所得的连翘HPLC对照指纹图谱对于连翘药材的鉴别具有很好的应用性[9]。
由此可得出结论本次实验所得的连翘的指纹图谱能够比较全面的反映连翘中各种化学成分的分布情况,且能从总体上反映连翘药材的质量和品质的优劣。
猜你喜欢
煤化工(2022年3期)2022-07-08
今日农业(2021年21期)2022-01-12
色谱(2021年7期)2021-06-07
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
今日农业(2020年16期)2020-09-25
中成药(2018年10期)2018-10-26
自动化学报(2016年8期)2016-04-16
中国资源综合利用(2016年10期)2016-01-22
青少年科技博览(中学版)(2015年7期)2015-08-12
