
新型α-二亚胺镍配合物的合成及其对乙烯聚合的催化
2018-09-18 09:41李向柳崔咪咪宋小雪侯彦辉
天津工业大学学报 2018年4期
杨 敏 ,李向柳 ,崔咪咪 ,孟 浩 ,姜 湃 ,宋小雪 ,侯彦辉
(1.河北工业大学 化工学院,天津 300130;2.天津工业大学材料科学与工程学院,天津 300387;3.天津工业大学省部共建分离膜与膜过程国家重点实验室,天津 300387)
自20世纪90年代开始,Brookhart等[1]报道了一系列α-二亚胺Ni和Pd配合物之后,这类后过渡金属催化剂的相关研究得到重视[2-11].作为继传统的Ziegler-Natta及茂金属催化剂之后发展起来的一类新型烯烃聚合催化剂,后过渡金属催化剂较前者有很多独特的优点.这类催化剂容易制备且成本不高;并且配合物本身结构较稳定,对水氧的敏感程度也较低;所合成的聚合物分子量分布较窄且结构可调控;特别之处是其能够催化唯一单体制备高支化或超支化结构聚合物,还可以催化烯烃与极性单体实现共聚[13].
众所周知,α-二亚胺类后过渡金属催化剂也有一些不容忽视的缺点,比如耐热性较差,这就极大的限制了它在工业化中的推进.因此,也吸引了很多课题组致力于催化剂性能的改善,并通过对配体结构的改变有效提高了其催化活性及热稳定性.比如,Guan等[5,14-16]报道合成了一系列二苯撑环烷烃骨架结构的α-二亚胺钯催化剂,考察了碳骨架结构的变化对于催化性能的影响.此外,还通过对苯胺对位取代基的改变,探究了取代基电子效应的不同对于催化效果的影响[16].伍青等[17]也设计合成了多种骨架结构不同的α-二亚胺镍催化剂,表明碳骨架位阻较大的催化剂催化活性较高,达到106g/(mol·h).而Long课题组[18-19]首先设计合成出的含大位阻二苯甲基取代基的α-二亚胺镍催化剂,甚至在温度达到100℃时都能保持很高的活性,展示出优异的热稳定性.Liu等[20-21]将二苯甲基取代基引入一侧苯胺的邻位或对位,制备得到了一系列结构不对称的α-二亚胺镍催化剂.探讨了苯胺取代基对于催化活性的影响,并从聚合结果证实其具备较高催化活性,甚至可以达到107g PE/(molNi·h).本课题组的翟飞帆等[22]合成了一类带有大位阻二苯甲基的二亚胺催化剂,在MAO存在时,70℃下仍能够以105g/(mol·h)的高活性催化降冰片烯均聚.另外,陈昶乐课题组[23]通过改变苯胺对位取代基的电子效应,发现吸电子基的存在对催化乙烯聚合有十分有利的影响.袁建超等[24-25]则合成了一类带有吸电子基的Br的α-二亚胺镍配合物,其催化乙烯聚合的热稳定性及所得产物的支化度有十分明显的提高.而现有的文献中大多为苯胺上取代基的改变,而关于改变碳骨架结构的报道相对较少.因此,本课题组设计合成一种新型的对称结构α-二亚胺镍催化剂Cat,通过对其苊醌骨架的调整,引入大位阻的苯环结构以及羟基官能团.之后,在助催化剂一氯二乙基铝(DEAC)作用下,探究聚合条件等因素对催化活性及聚合产物熔点及分子质量等的影响.
1 实验部分
1.1 实验原料及试剂
二氯甲烷,分析纯,天津市科锐思精细化工有限公司产品,氩气保护下加氢化钙干燥并回流,蒸出后使用;甲苯,分析纯,天津市华东试剂产品;正己烷、分析纯,天津市科锐思精细化工有限公司产品;四氢呋喃,分析纯,天津市科锐思精细化工有限公司产品,氩气保护下加入金属钠回流,用前蒸出;无水乙醇,分析纯,天津恒山化工科技有限公司产品,直接使用;石油醚、乙酸乙酯,均为分析纯,直接使用;酸化乙醇溶液,体积分数为10%,自配,直接使用;苊(98%),上海迈瑞尔化学技术有限公司产品;N,N-二甲基甲酰胺(DMF),分析纯,天津市光复精细化工研究所产品;N-溴代丁二酰亚胺(NBS,99%),上海迈瑞尔化学技术有限公司产品;重铬酸钠(99%),天津市安吉瑞化工有限公司产品;乙酸,分析纯,上海迈瑞尔化学技术有限公司产品;4-羟甲基苯硼酸(99%)、2,6-二异丙基苯胺(99%),安耐吉化学产品;乙烯(聚合级)、氩气(99.9%),空气化工(天津)有限公司产品,直接使用区;一氯二乙基铝(DEAC),1.0 mol/L正己烷溶液,百灵威科技有限公司产品,直接使用.
1.2 测试与表征
采用Brucker公司Vector-22型FT-IR光谱仪对配体L和催化剂Cat进行表征(KBr压片法,扫描范围为 4000~400 cm-1);常温下采用 Bruker DMX400 型核磁共振仪对配体及配合物进行核磁表征,溶剂为氘代氯仿;采用Flash EA 1112型全自动元素分析仪对催化剂进行元素分析.采用DSC-Diamond型(Perkin-Elmer公司)差示扫描量热仪测定聚合物熔点,N2气氛下,测试范围0~150℃(升温速率:10 K/min),取二次升温结果,得到聚合物熔点Tm及熔融焓ΔHf,结晶度χc=其中为完全结晶的熔化热,文献参考值为276.14 J·g-1[23];聚乙烯分子质量及分子质量分布采用英国Polymer lab公司PL-GPC220于140℃温度下进行测定,采用1,2,4-三氯苯作为溶剂,样品浓度为1 mg/mL,流速为1.0 mL/min;聚乙烯产物的13C-NMR采用Brucker公司DMX-400型核磁共振仪在温度为120℃下,以氘代邻二氯苯为溶剂进行测试.
1.3 配体及催化剂的合成
1.3.1 N,N-二(2,6-二异丙基苯基)-5,6二溴苊烯-1,2-二亚胺(C)的制备
在冰浴下将溶有 DMF 的 NBS(6.4 g,0.036 mmol)混合物逐滴滴入溶有DMF的苊(2 g,12.96 mol)中.滴加完毕室温下搅拌过夜,次日抽滤得粗品,乙醇重结晶得白色固体4,5-二溴苊(A).将化合物A(0.64 g,2 mmol)与重铬酸钠(3.5 g,11.6 mmol),溶于35 mL 乙酸中,80℃下搅拌反应2 h.冷却至室温后抽滤,水洗,烘干得化合物 5,6-二溴苊烯-1,2-二酮(B).将化合物 B(1.3 g,3.8 mmol)和 2,6-二异丙基苯胺(2.2 mL,11.4 mmol)溶于甲苯中,加入适量对甲基苯磺酸,加热回流2h,蒸发溶剂后冷却至室温,柱层析分离提纯得N,N-二(2,6-二异丙基苯基)-5,6 二溴苊烯-1,2-二亚胺(化合物C)为0.50 g,产率20%.
1.3.2 N,N-二(2,6-二异丙基苯基)-5,7-二(4-羟甲基苯基)苊烯-1,2-二亚胺(L)的制备
取化合物 C(0.5 g,0.305 mmol),对羟甲基苯硼酸(0.3 g,1.9 mmol)和无水碳酸钾(0.8 g,5.71mmol)于500 mL茄形瓶中,之后加入适量的三苯基磷钯催化剂,置换气体,在氩气保护下用注射器分别加入50 mL经氢化钙干燥的THF与适量蒸馏水,加热回流10 h.反应结束,冷却至室温,用二氯甲烷进行萃取,无水硫酸镁干燥,滤液旋干,最后通过柱层析分离提纯得纯品(L)10.5 g,产率 25%.1H NMR(400 MHz,CDCl3):δ 7.38-7.33(d,8H),δ 6.97-6.82(m,10H),δ 4.59(s,4H),δ3.20-3.13(m,4H),δ 1.35(d,2H),δ 1.33(d,12H),δ 1.12-1.10(d,12H).C50H52Br2N2NiO2(712.96)的元素分析:理论值(%):C,84.23;H,7.35;N,4.49;实测值(%):C,84.55;H,7.22;N,4.68.
1.3.3 N,N-二(2,6-二异丙基苯基)-5,7-二(4-羟甲基苯基)苊烯-1,2-二亚胺溴化镍(Cat)的制备
所有操作均在无水无氧条件下进行,反应体系经氩气3次置换.在100 mL三口瓶中加入Ni(DME)Br2(1 mmol)和20 mL二氯甲烷.取1 mmol配体 L,并用10 mL二氯甲烷溶剂将其充分溶解后,将溶液缓慢滴加至三口瓶中.40℃下回流搅拌反应10 h,除去溶剂,加入适量正己烷洗涤沉降多次,直至上层液无色.将溶剂进一步抽干,得到干燥的橙红色催化剂Cat粉末,产率为86%.C50H52Br2N2NiO2(931.48)的元素分析:理论值(%):C,64.47;H,5.63;N,3.44;实测值(%):C,64.75;H,5.42;N,3.58.其中,(DME)NiBr2参考相关文献[24]制备,得到淡黄色粉末,元素分析C4H10Br2NiO2:实测值(%):C,15.40;H,3.58;理论值(%):C,15.57;H,3.27.
1.4 乙烯聚合
所有实验操作均使用Schlenk标准技术在氩气气氛保护下进行.将装有搅拌磁子的反应容器(三口瓶或高压反应釜)在进行真空-氩气置换3次后,充入乙烯气体,并保持一定压力.设定温度至反应所需,在搅拌下用注射器顺次加入甲苯溶剂、催化剂溶液、助催化剂一氯二乙基铝(DEAC),迅速充压至反应压力,开始计时.反应结束,切断乙烯供给,降温并泄压,将反应液缓慢倾倒至10%酸化乙醇中进行终止.聚合产物经乙醇、水交替洗涤,抽滤后60℃下真空干燥至恒重,称量并计算活性见下式:
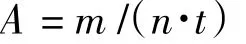
式中:A为催化剂的活性(g/(mol·h));m为产物质量(g);n为催化剂用量(mol);t为聚合时间(h).
2 结果与讨论
2.1 配体及配合物的合成
图1所示为配体L及其对应镍配合物Cat的具体合成路线.
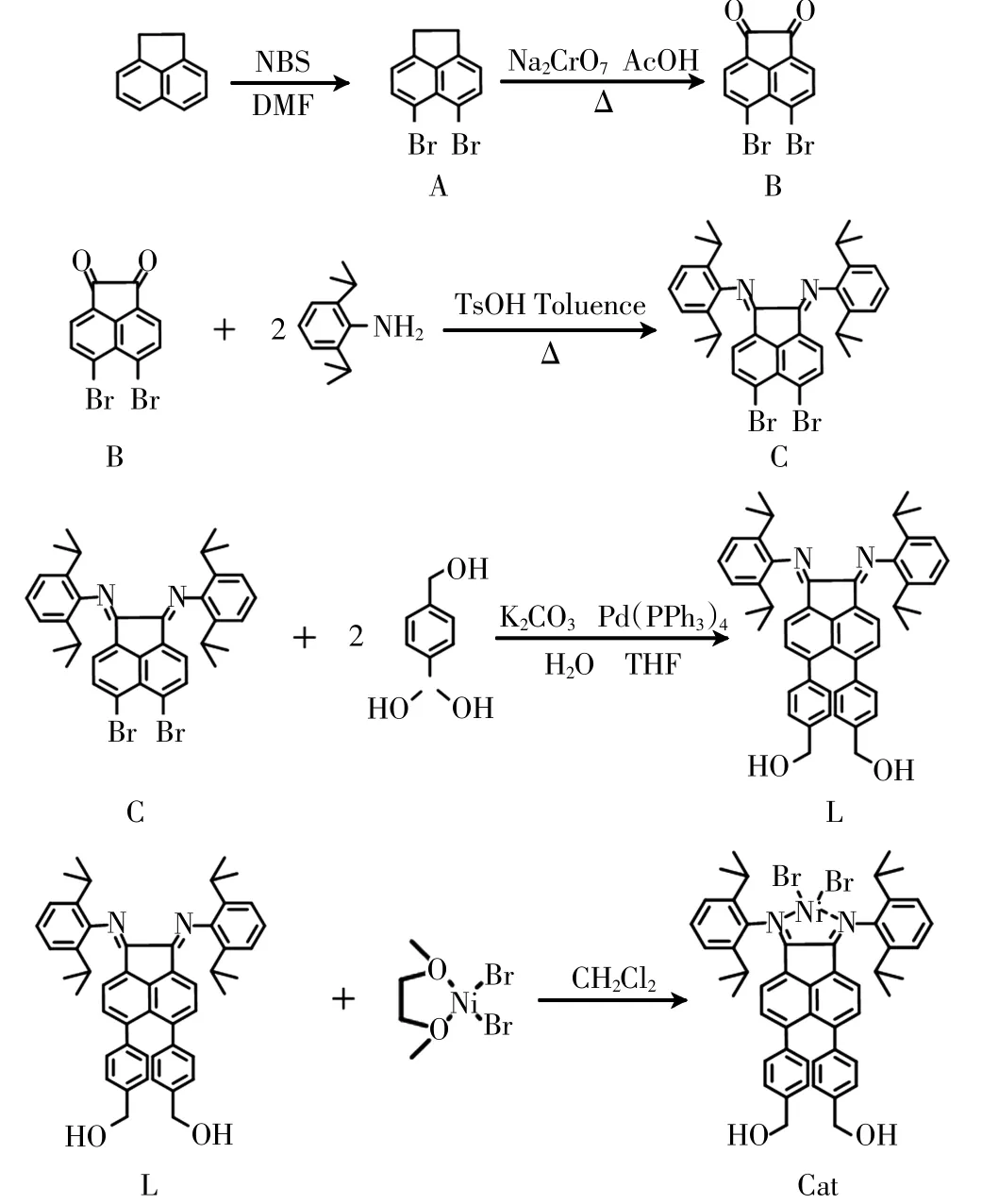
图1 配体L及配合物Cat的合成Fig.1 Synthesis of ligands L and complex Cat
鉴于配合物Cat中金属镍有很强的顺磁性,利用NMR表征无法得到完全理想的谱图,故借助红外吸收光谱对其表征,结果如图2所示.
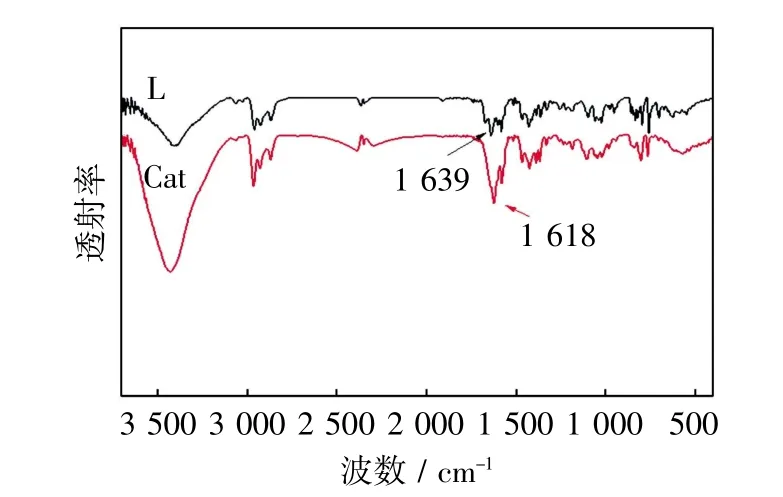
图2 配体L和配合物Cat的红外谱图Fig.2 FT-IR spectra of ligand L and complex Cat
由图2可见,配体L的红外光谱在1639 cm-1附近有很强的C=N伸缩振动峰.而配合物Cat的红外光谱中C=N伸缩振动峰(1618 cm-1)相比配体而言明显向低频方向发生了移动.配体L与配合物Cat在其余位置的吸收峰基本一致,初步说明金属Ni与配体中N原子确实发生了配位,这也与元素分析的结果相一致.
2.2 乙烯聚合结果
以配合物Cat为主催化剂,在助催化剂一氯二乙基铝的作用下催化乙烯进行常压或高压聚合.其中,聚合温度、铝镍比(n(Al)/n(Ni))以及乙烯压力对其催化乙烯聚合性能的影响结果分别见表1、表2及图3.

表1 聚合温度对催化性能的影响Tab.1 Effect of polymerization temperature on catalytic propreties
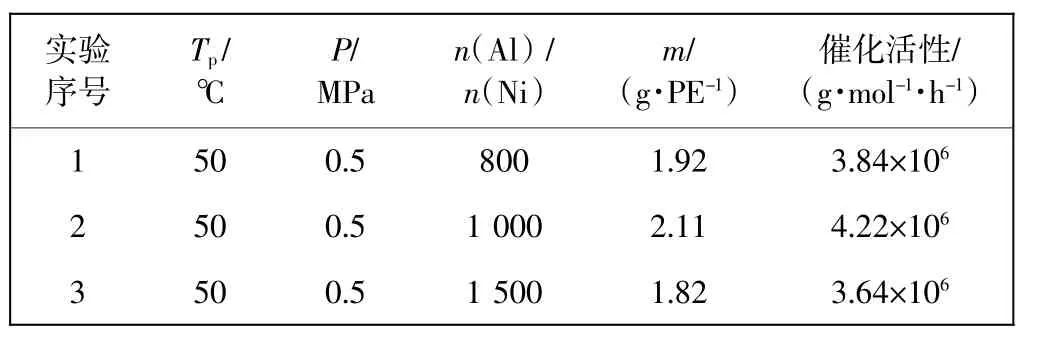
表2 铝镍比对聚合活性的影响Tab.2 Effect of Al/Ni on activity
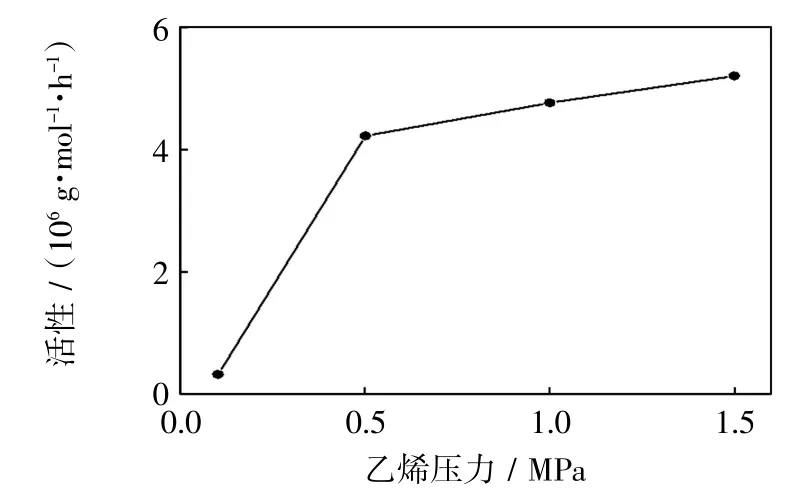
图3 乙烯压力对催化活性的影响Fig.3 Effect of ethylene pressure on activity
由表1可知,在乙烯压力为0.5 MPa的聚合条件下,随着聚合温度从30℃升高至80℃,其催化乙烯聚合的活性呈现出先升高后降低的变化趋势,并在50℃条件下达到最高活性为4.22×106g/(mol·h).分析其原因为,温度较低时的链增长速率较为缓慢,故温度的升高有利于反应的加速进行,催化活性呈现升高的变化趋势.而当温度超过一定范围,较高的温度环境则会影响催化剂活性中心的稳定性,从而导致催化剂的分解和部分失活,催化活性又随之下降.同时,当聚合温度升高至80℃时,虽然其催化活性呈现降低的变化趋势,但仍可以达到1.42×106g/(mol·h),反映出了催化剂Cat良好的热稳定性.另外,随着聚合温度由50℃增加至70℃,聚合物对应的数均分子量由143 kg/mol降低至125 kg/mol.这是由于聚合物分子质量的大小受链增长和与之相竞争的各种链转移反应的共同影响,较高的温度会导致链转移的活化能大于链增长的,也就使得链转移的速率相对增加更快,相应聚合物的分子质量降低.
由表2可知,当保持聚合温度和压力不变时(50℃,0.5 MPa),催化活性随着铝镍比增加展现出先增加再减低的趋向.这主要归因于助催化剂DEAC在催化体系中的作用除了能够清除反应杂质之外,还能够与主催化剂发生单烷基化反应,使得配合物被夺去电子成为缺电子的阳离子活性中心,有利于单体插入聚合.故铝镍比较低时,活性中心的形成可能并不充足,一定范围内,助催化剂用量的增加有利于提高聚合活性.而DEAC过多极可能导致聚合物链向其自身发生转移,反而降低聚合活性.
由图3可知,随着聚合压力从0.1 MPa提高到1.5 MPa,催化活性一直呈现出升高的趋势,并且增长趋势逐渐趋缓.原因是压力的增大有利于乙烯单体在溶剂中的溶解,增加活性中心与乙烯单体碰撞的几率,催化活性增加.而当压力达到一定范围,聚合速率增大的同时,聚合体系的黏度也在随之增加,会影响乙烯单体在甲苯溶剂中的溶解与扩散,活性中心以及单体容易被已生成的聚合物包埋,这两方面因素促使催化活性增加不明显.
2.3 聚乙烯的相关表征
本文选取不同聚合温度下制备的聚乙烯样品,采用13C-NMR对其进行表征,图4所示为产物的核磁谱图.
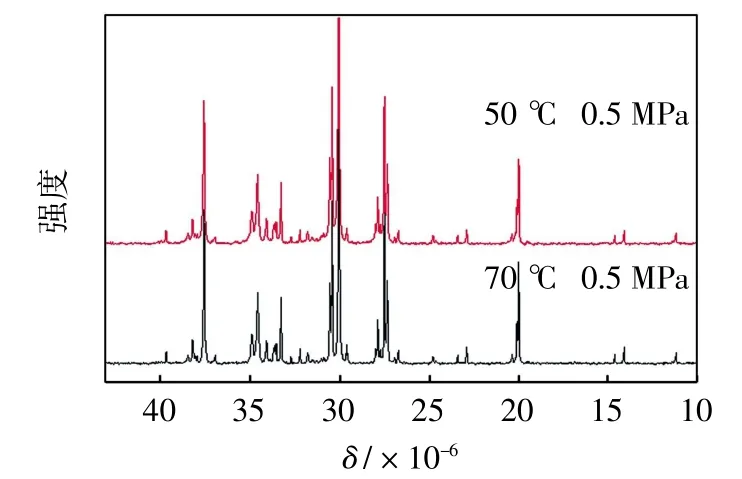
图4 0.5 MPa下所得聚乙烯产物的13C-NMR谱图Fig.4 13C-NMR spectra of polyethylenes synthesized at 0.5 MPa
由图4可以清晰地看出,所得聚乙烯具有复杂的支链结构,包括甲基、乙基等短支链,也包括戊基、己基及更长的长支链,并且当聚合温度由50℃增加至70℃时,能观察到一些支链所对应峰强度的增加.根据相关文献[28]计算各产物的总支化度及各支链所占比例,如表3所示,能够得出与图4的观察结果相一致的结论.表3中数据表明,当聚合温度为50℃时,其总支化度为125个支链/1000 C,其中甲基支链占66.41%,其余长支链占5.79%;而70℃时对应的支化度为135个支链/1000 C,甲基支链所占比例降低至60.00%,长链比例增加至7.71%.其可能原因是,反应的温度升高使得“链行走”相对于链增长的速率增加,从而使支化度总体上提高.同时,由于聚乙烯产物有较高的支化度,其DSC曲线中并未出现明显的熔融峰.

表3 聚乙烯产物的支化度分布Tab.3 Branching degrees of selected polyethylene samples
3 结论
本文通过对苊醌碳骨架上取代基的改变,与2,6-二异丙基苯胺反应制备得到大位阻骨架的α-二亚胺镍配合物Cat,并通过红外及元素分析等测试手段对配合物结构进行了表征.以此α-二亚胺镍配合物作为主催化剂,一氯二乙基铝作为助催化剂催化乙烯聚合得到分子量分布在2.2左右,支化度约为130个支链/1000 C的支化聚乙烯产物.结果表明,苊醌碳骨架上大空间位阻取代基的存在使得该催化剂能保持良好的热稳定,在80℃下仍可展现较高活性.另外,随着聚合温度的增加,其聚合活性呈现先升高后降低的变化趋势,并在50℃条件下达到最高值;聚合温度的由50℃增加至70℃时,聚乙烯对应的数均分子质量由143 kg/mol降低至125 kg/mol,其总支化度由125个支链/1000 C增加至135个支链/1000 C,并且相应短支链的比例降低,长支链比例增加.随着铝镍摩尔比由800增加至1500,其催化乙烯聚合的活性先升高后降低,但变化范围不大.而乙烯压力的增加,会导致其催化活性呈现随之升高的变化趋势,但其升高幅度会逐渐趋于平缓.
猜你喜欢
橡塑技术与装备(2022年9期)2022-09-05
检验医学(2022年2期)2022-03-14
合成化学(2022年1期)2022-02-19
冶金能源(2022年1期)2022-02-18
核科学与工程(2021年4期)2022-01-12
农业机械学报(2021年10期)2021-11-09
医药前沿(2018年25期)2018-08-20
农业机械学报(2018年7期)2018-07-30
中华胃食管反流病电子杂志(2017年2期)2017-10-27
北京航空航天大学学报(2014年1期)2014-12-19
