
Ruddlesden-Popper结构杂化非本征铁电体及其多铁性∗
2018-09-06 07:32刘小强吴淑雅朱晓莉陈湘明
物理学报 2018年15期
刘小强 吴淑雅 朱晓莉 陈湘明
(浙江大学材料科学与工程学院,杭州 310027)(2018年2月9日收到;2018年3月13日收到修改稿)
1 引 言
铁电体是指具有可被电场翻转的自发极化的极性材料,广泛应用于压电元件、非易失性存储器、固态制冷以及太阳能电池等方面[1,2].磁电多铁性材料则是指同时具有铁电性和(反)铁磁性,并具有磁电耦合效应的材料,其在自旋电子学、微弱磁场探测以及低功耗多态高密度存储等领域有着广阔的应用前景[3−5].目前,单相多铁性材料还面临种种困难和挑战.这是因为传统铁电性来源于金属离子与周围氧离子之间杂化形成的赝扬-特勒(pseudo Jahn-Teller)效应,而该效应要求金属离子一般是具有空d轨道的离子,如Ti4+等.相反,铁磁或反铁磁性则要求具有非空d轨道的离子.从电子构型来看,铁电性和(反)铁磁性是相互排斥的[6].因此,在多铁性材料研究初期,只发现了Pb(Fe1/2Nb1/2)O3等极少数单相化合物具有低温多铁性[5].直到2003年,BiFeO3外延薄膜制备与磁电耦合的表征以及TbMnO3中巨大磁电耦合效应的发现,单相多铁性材料才真正引起凝聚态物理与材料科学领域的普遍关注[5,7−15].以BiFeO3为代表的第I类多铁性材料虽然具有室温铁电和(反)铁磁性共存与大铁电极化的优点,但磁电耦合效应微弱.而正交结构TbMnO3等第II类多铁性材料的铁电极化源自特定的磁结构,故而显示出很强的磁电耦合效应.其问题是铁电极化微弱,比正常铁电体低4个数量级,更严重的问题是其铁电与磁转变温度远低于室温[4,5,8].
为了避免电子构型排斥的出现,Young等[16]提出了利用非本征铁电性来解决室温多铁性材料面临的问题.在BaTiO3等经典铁电材料中,铁电极化源于TiO6八面体中Ti4+偏离氧八面体中心的位移,其自发极化的出现能够完全表征从顺电到铁电相变的所有对称性变化,这是本征铁电性.本征铁电性是由赝扬-特勒效应引起的,需要与磁性相排斥的电子构型,故难以形成单相多铁性材料.而非本征铁电体中的自发极化是由其他非极性畸变诱导出的,故自发极化的出现不能够完全描述相变过程中的所有对称性变化.同时,其相变是由非极性畸变引起的,因此其电子构型没有上述本征铁电体的要求,这就避免了与磁性电子构型的排斥,故有望容易获得单相多铁性材料.而最近提出的杂化非本征铁电性(hybrid improper ferroelectricity,HIF)不仅能更加容易地实现铁电性和磁性的共存,还有望实现内禀的电控磁特性.因此,可望获得具有强磁电耦合的室温多铁性材料[17].
本文在阐述HIF物理模型的基础上,总结了有关Ruddlesden-Popper(R-P)结构杂化非本征铁电体及其多铁性的主要研究进展,分析了其面临的主要挑战与发展前景.
2 HIF的物理模型
在简单钙钛矿ABO3中,当A,B位离子的半径合适时,即其许容因子(tolerant factor)接近1时(如BaTiO3,室温空间群为P4mm),BO6八面体不会出现旋转(rotation)或倾侧(tilt).而随着许容因子的降低,为了保持结构的稳定性,BO6八面体将出现旋转或倾侧,如CaTiO3(室温空间群为Pbnm).特别是在钙钛矿及类钙钛矿中,如果同时存在a0a0c+和a−a−c0两种氧八面体的倾转模式,将导致A位离子出现反铁畸变位移,且相邻层的位移方向相反[16−19].在简单钙钛矿ABO3中,由于A位离子是相同的,相邻层的反铁畸变位移相互抵消,最终形成非极性的结构.最典型的例子便是CaTiO3,其结构为非极性正交Pbnm相.而在A位离子有序的双钙钛矿结构中,相邻层A位离子不同,其反铁畸变位移不一致,单胞内的位移不能完全抵消,从而形成了铁电位移[19,20].典型的例子是NaLaMnWO6,其结构为极性的单斜P21相[21].以上形成的就是HIF,而之所以称之为杂化,是因为两种氧八面体的倾转模式可以在不同的温度下冻结[17].从以上的分析可知,HIF仅出现在A位有序的钙钛矿材料中,而A位有序在钙钛矿化合物并不常见,这就极大地限制了HIF.
层状钙钛矿结构是钙钛矿结构的重要拓展,其主要包含以下3类[22]:Dion-Jacobson结构(D-J结构,通式为R-P结 构(通 式 为)和Aurivillius结构(通式为具体结构如图1所示.层状钙钛矿是由n层钙钛矿和单层岩盐层(D-J结构)或双层岩盐层(R-P结构)或Bi2O2层(Aurivillius结构)沿着[001]方向排布而成.当n=∞时,层状钙钛矿转变为钙钛矿结构.岩盐层或Bi2O2层的插入打破了原来的对称性,可以移除在B位上的对称中心,从而有望形成极性对称群.研究者们很早就在Aurivillius结构中发现了铁电材料,如Bi4Ti3O12,Bi2SrTa2O9等.虽然该类材料的铁电相变中也涉及到氧八面体的旋转,且其与极性对称模存在耦合,但相变是由极性模主导的[16].因此,从严格意义上讲,此类材料不属于HIF.而关于D-J结构的铁电性问题,虽然有第一性原理计算预测某些体系确实存在HIF,但到目前为止,仅在含Bi3+的ABiNb2O7(A=Rb,Cs)中获得了确切的实验证据[23].对于R-P结构,目前已有大量的计算和实验结果表明HIF的存在[24−31].因此,本文主要关注R-P结构的HIF.
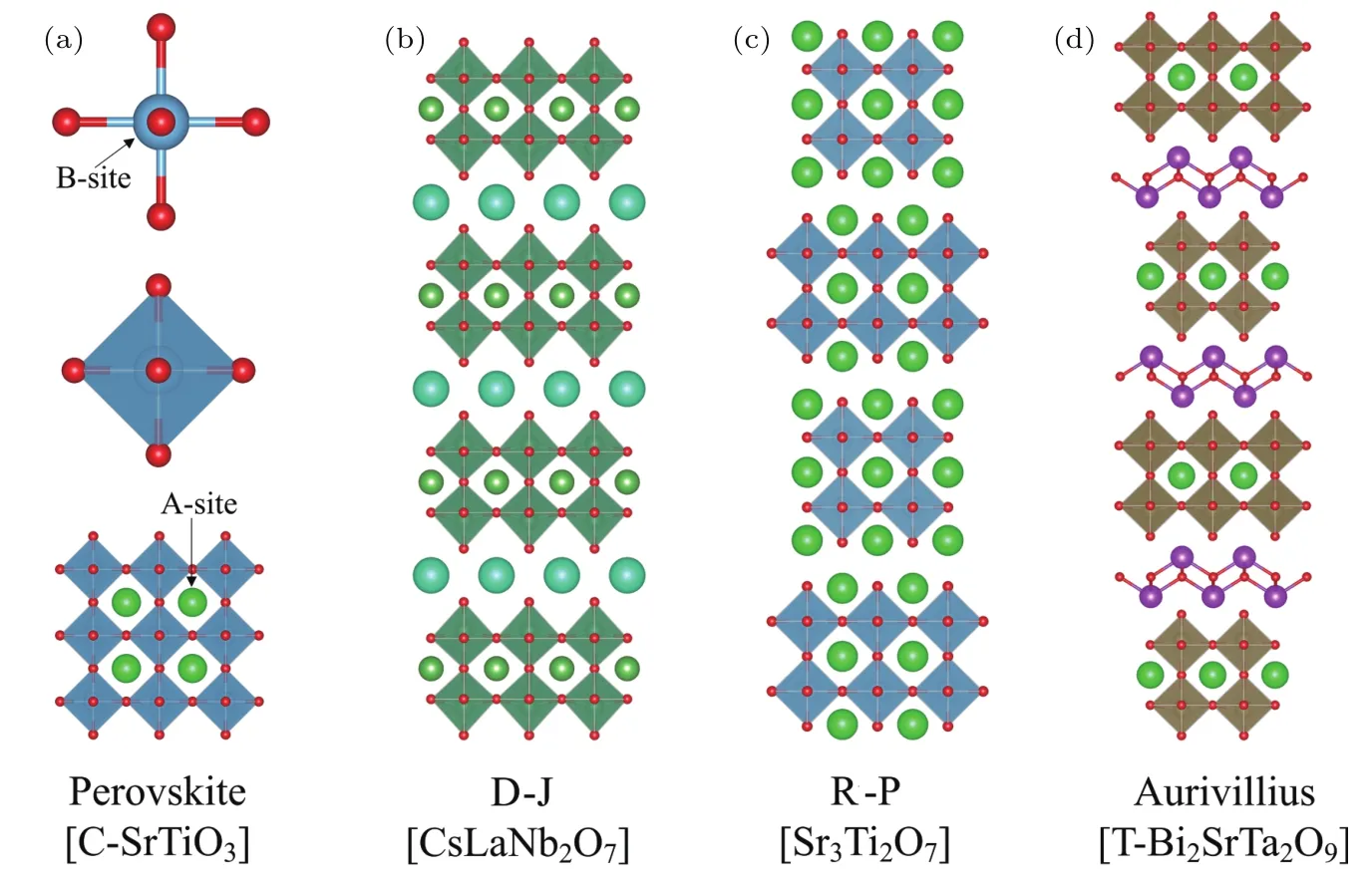
图1 钙钛矿及典型层状钙钛矿结构示意图 (a)简单钙钛矿结构立方SrTiO3;(b)n=2,D-J结构CsLaNb2O7;(c)n=2,R-P结构Sr3Ti2O7;(d)n=2,Aurivillius结构四方Bi2SrTa2O7[22]Fig.1.Representative structures of perovskite and layered perovskites:(a)Cubic-SrTiO3with simple perovskite structure;(b)CsLaNb2O7with n=2 D-J structure;(c)Sr3Ti2O7with n=2 R-P structure;(d)tetragonal Bi2SrTa2O7with n=2 Aurivillius structure[22].

图2 R-P结构从(a)顺电相到(b)铁电相的对称模分解以及(c)铁电相中每层沿a轴的反铁畸变位移(X)及总的铁电极化(Ptotal)示意图[16]Fig.2.Symmetry mode decomposition of the(a)paraelectric to(b)ferroelectric structure in R-P A′2AB2O7,and(c)the representation of antiferrodistortion displacements(X)at every layer and the total ferroelectric polarization(Ptotal)in the structure[16].
HIF是由面内旋转a0a0c+和面外倾侧a−a−c0两种氧八面体的倾转模式引起的[17].在R-P结构中,其分别对应四方顺电相中布里渊区X点上的和两个对称模,而这两个对称模的耦合导致极性模的出现,从而诱导出A位离子的极性位移,形成正交铁电相,具体如图2所示[16−18].需要强调的是,正交铁电相的建立仅需和两个对称模,不需要极性模的参与.因此,HIF中铁电性是由氧八面体倾转诱导的,是二阶铁电序.在n=2的R-P结构中,A位离子出现的反铁畸变位移如图2(c)所示,在钙钛矿层中沿着−a方向,在岩盐层中则沿着a方向.在一个结构单元中,两个位移沿着a方向,一个位移沿着−a方向,因此,即使在A位离子相同或完全无序的情况下,反铁畸变位移仍然不能完全抵消,形成总的铁电极化.即在n=2的R-P结构中,A位离子有序不再是获得HIF的前提,这极大地扩展了HIF材料的范围.事实上,在n为偶数的R-P结构中均可出现HIF,但由于一个单胞中只有一个反铁畸变位移没有抵消,随着n的增加,晶胞体积增加,故总的极化值降低.因此,n=2的R-P结构是最具有研究价值的[16−18].
按照朗道相变理论,HIF在无外加电场下的自由能F可以表示为[32]
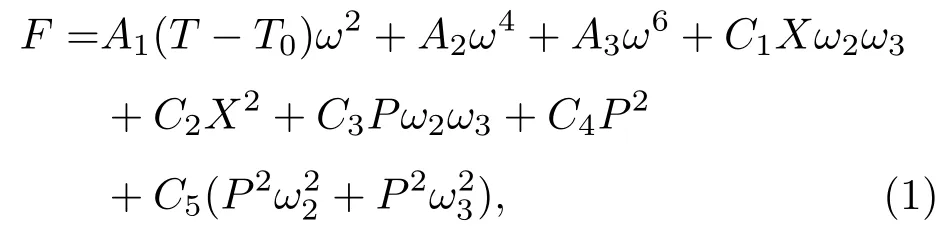
HIF中的铁电极化是由氧八面体的面内旋转和面外倾侧耦合导致的.因此,铁电极化的翻转必然导致氧八面体的倾转反转.如果在氧八面体中心是磁性元素(如Mn4+,Fe3+等),其通过超交换作用形成(倾斜的)反铁磁长程有序.由于铁电极化是由A位离子的反铁畸变位移引起的,与氧八面体中的离子无关,只要在氧八面体中加入磁性离子形成长程有序,就可形成单相多铁性材料.另外,由于超交换作用对于O2−—磁性离子—O2−键角十分敏感,在氧八面体倾转反转时,该键角必然会随之变化,从而导致材料的磁学性能发生变化,这是一个典型的电控磁特性[16−18].因此,HIF可能是获得具有电控磁特性多铁性材料的有效途径,可望同时实现室温大铁电极化与强磁电耦合.
综上所述,HIF是指在具有钙钛矿结构单元的金属氧化物中由氧八面体面内旋转和面外倾侧耦合而诱导出的二阶铁电序,其有望在强磁电耦合多铁性材料中获得重要应用,并将极大地拓展铁电体物理学的内涵和外延.
3 R-P结构杂化非本征铁电体的最新进展
早在2008年,Bousquet等[33]在研究PbTiO3/SrTiO3超晶格的铁电性时,发现当把两层的比例从9/3降到2/3时,其性能从本征铁电性转变为非本征铁电性.而该非本征铁电性是由两个氧八面体倾转诱导的,这应该是HIF的研究肇始.

图3 (Ca,Sr)3Ti2O7单晶的铁电性 (a)和(b)空间群为A21am的(Ca,Sr)3Ti2O7单晶晶体结构;(c)和(d)为Ca2.46Sr0.54Ti2O7单晶的(001)解理表面照片和室温环形差分干涉衬度照片;(e)Ca3−xSrxTi2O7(x=0,0.54,0.85)单晶沿[110]方向的电滞回线;(f)IP-PFM的配置示意图[24]Fig.3.Planar electric polarization of Ca3−xSrxTi2O7single crystals at room temperature:(a)and(b)Crystallographic structure of Ca3−xSrxTi2O7with the orthorhombic A21am space group(the layered perovskite structure consists of a perovskite(P)block and a rock-salt(R)block);(c)photographic and(d)circular dif f erential interference contrast image of a cleaved(001)surface of a Ca2.46Sr0.54Ti2O7single crystal;(e)ferroelectric hysteresis loops of Ca3−xSrxTi2O7(x=0,0.54,0.85)single crystal along[110]orientation;(f)schematic picture of our IP-PFM measurement[24].
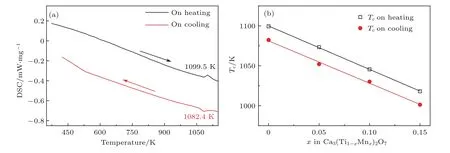
图4 (a)Ca3Ti2O7陶瓷的DSC信号在升温和降温的条件下随着温度的变化;(b)Ca3(Ti1−xMnx)2O7陶瓷的吸热或放热峰温度Tc随着成分的变化规律,其中实线为拟合结果[25]Fig.4.(a)Temperature dependence of DSC signs for Ca3Ti2O7ceramics during the heating and cooling cycles;(b)temperature Tcof endothermic or exothermic peak for Ca3(Ti1−xMnx)2O7ceramics,and solid lines are the linear f i tting results[25].
2011年Benedek和Fennie[17]利用第一性原理预测了Ca3M2O7(M=Ti,Mn)块体中存在HIF,并提出了HIF的概念和物理模型,但在很长时间内没有得到实验确认.直到2015年,Oh等[24]成功地生长出了(Ca,Sr)3Ti2O7单晶,并在室温下利用PUND(positive-up-negative-down)方法测得了电滞回线,具体结果如图3所示.首先利用X射线衍射(X-ray dif f raction,XRD)拟合出晶胞参数,然后由晶胞参数计算出正交度,最后通过正交度确定Ca3−xSrxTi2O7在0 6 x 6 0.9是正交相[24].当置换量高于这个范围,则为四方相.图3(a)和图3(b)分别给出了a0a0c+和a−a−c0两个氧八面体倾转模式的示意图,并同时用红色和蓝色箭头表示了A位(Sr,Ca)离子的反铁畸变位移.图3(c)和图3(d)则分别给出了Ca2.46Sr0.54Ti2O7单晶的(001)面的表面形貌和环形差分干涉衬度照片,可以明显看出该晶体中存在正交孪晶.图3(e)则给出了Ca3−xSrxTi2O7(x=0,0.54,0.85)单晶在室温下测得沿[110]方向的电滞回线.从图3(e)可知,晶体中确实存在可翻转的自发极化,其剩余极化最高可达8µC/cm2,这首次从实验上确认了HIF的存在.同时,其矫顽场约在150 kV/cm,远低于计算预期,这应该是实际翻转路径与理论预测不一致导致的.另外,还测试了沿[001]方向的电滞回线,结果为一条通过原点的直线,说明晶体沿着c轴方向上没有可翻转的自发极化,这进一步验证了该材料的铁电性确实起源于A位离子未抵消的反铁畸变位移.为了进一步探索其铁电畴结构,使用如图3(f)所示的面内压电响应力显微镜(in-plane piezo-response force microscopy,IP-PFM)来表征材料中的铁电畴,发现了丰富的头对头的导电畴壁和尾对尾的绝缘畴壁.Huang等[28]则利用原位电子衍射结合介电响应在Ca3−xSrxTi2O7(x=0.915—1)附近发现了一个新的四方P42/mnm相.
Liu等[25]随后利用标准固相反应法制备了Ca3(Ti1−xMnx)2O7(x=0,0.05,0.10,0.15)陶瓷,也在室温下使用PUND方法获得了非线性电滞回线.相对于单晶材料而言,陶瓷的剩余极化值小一个数量级,最大极化值约为0.6µC/cm2,但两者矫顽场基本一致.为了研究材料的居里温度,通过高温差示扫描量热法(dif f erential scanning calorimetric,DSC)测量在Ca3Ti2O7陶瓷升温至1099.5 K附近观察到一个吸热峰,同时降温至1082.4 K附近出现了一个放热峰(图4(a)).同样,在其他3个成分中也发现了吸热和放热峰,其具体温度如图4(b)所示.按照相变分类,存在热滞的相变为一级相变.由于这些温度存在线性关系,可通过拟合推测出Ca3Mn2O7的相变温度,约为550 K,与文献[34]报道相近.Liu等[25]利用上海光源的原位高温XRD测定了其相变温度以上的晶体结构.高温XRD的拟合结果表明,Ca3Ti2O7陶瓷在1173 K为四方I4/mmm相,这与理论预测一致[17].Li等[29]也利用高温DSC研究了Ca3−xSrxTi2O7(x=0.1,0.2,0.3,0.4)陶瓷的相变温度随着Sr2+置换量的变化规律.研究结果表明,该系列陶瓷也呈现出一级相变特征,且其相变温度随着Sr2+置换量的增加而线性下降,外推的结果与Huang等[28]的结果一致.
Li等[26]利用脉冲激光沉积首次在(110)Sr-TiO3基板上成功地生长了Ca3Ti2O7薄膜,并用一系列微结构表征证明了[001]Ca3Ti2O7//[001]SrTiO3和 [110]Ca3Ti2O7//[¯110]SrTiO3外 延 关 系.铁电测试表明,其自发极化沿着面内的a轴.令人特别感兴趣的是,薄膜的矫顽场仅为5 kV/cm,大大低于单晶和陶瓷的矫顽场.第一性原理计算表明,块体和薄膜的矫顽场应该一致,而薄膜中如此低的矫顽场应该归结于薄膜中的非完美晶格.

图5 多晶Sr3Sn2O7样品的室温正交孪晶结构和铁电极化 (a)抛光表面在透射模式下的偏振光显微照片;(b)室温下的电滞回线;(c)285 K下的XRD图谱;(d)和(e)铁电a−a−c+畸变模式在c和b轴方向投影[30]Fig.5.Orthorhombic twin domains and switchable electric polarization of a polycrystalline Sr3Sn2O7specimen at room temperature:(a)Polarized optical microscope images of the polished surface in a transmission mode;(b)electric polarization(P)and compensated current(I)versus electric f i eld(E)hysteresis loop by a PUND method;(c)XRD pattern of Sr3Sn2O7at 285 K;(d)and(e)c-direction and b-direction views of the ferroelectric a−a−c+distortion in Sr3Sn2O7[30].
Wang等[30]还在Sr3Sn2O7陶瓷中观察到HIF,具体结果如图5所示.首先利用偏光显微镜的透射模式观察多晶薄片,观察到了如图5(a)所示的明暗相间衬度,这些衬度是由正交孪晶结构引起的.随后利用PUND方法测试了室温电滞回线(图5(b)),其剩余极化在0.1µC/cm2左右,矫顽场在200 kV/cm左右.XRD的分析结果表明,衍射结果既可以用极性A21am相拟合,也可以用非极性Amam相拟合,但结合前面的电滞回线结果,则应该用极性相拟合.图5(d)和图5(e)则分别给出了铁电a−a−c+倾转模式在c和b轴方向投影.同时还用透射电镜的暗场相模式确认了材料确实是由极性相组成的.
4 R-P结构多铁性材料
HIF中铁电位移是由A位离子未完全抵消的反铁畸变位移引起的,而磁性则由氧八面体中心磁性元素的超交换引起的.因此,铁电和磁性共存的多铁性材料应该容易获得.Pitcher等[31]率先在铁基R-P结构中获得了极性相和弱铁磁性共存的室温多铁性材料,并在低温下测得了线性磁电耦合系数,具体结果如图6所示.为了获得室温多铁性材料,选取了反铁磁奈尔温度较高的铁基材料作为改性基体,然后通过在A位引入半径较小的离子,降低材料的许容因子,从而引入氧八面体的倾转,达到获得HIF的目的.图6(a)给出了(1 − x)(Sr0.4Ca0.6)1.15Tb1.85Fe2O7−xCa3Ti2O7(0 6 x 6 0.30)的结构和磁性相图,反铁磁奈尔温度随着x的增加而缓慢降低,而铁电极性相的相变温度随着x的增加而增加,在中间区域里出现了极性相和弱铁磁相共存的现象.图6(b)给出了该区域的放大图,从图中可知,室温多铁性存在于0.13 作为HIF在多铁性中应用的模型材料Ca3Mn2O7,其电滞回线一直没有报道,而其磁电耦合效应却异常地高.最近,Gao等[27]成功地生长了Ca3Mn2−xTixO7(x=0,0.1,1.0,1.5,2.0)单晶,发现Mn含量较高的Ca3Mn2−xTixO7(x=0,1.0)单晶在77 K可测得可信的线性电滞回线,即不存在可翻转的自发极化.而在Ti含量较高的成分中则可以获得非线性的电滞回线,即存在可随电场翻转的自发极化.利用暗场透射电子显微镜观察其铁电畴,发现Ca3Mn2O7单晶中存在大量的90◦孪晶畴与铁电畴交错排布,而这些孪晶的存在阻碍了铁电畴的翻转.因此,该材料中虽然存在铁电畴,但铁电畴翻转十分困难,故只能测出线性的电滞回线,其磁电耦合效应也很小.而在Ca3Ti2O7中,不存在上述的孪晶畴与铁电畴交错排布的现象,铁电畴容易翻转,故可获得完美的电滞回线. 图6 多晶(1−x)(Sr0.4Ca0.6)1.15Tb1.85Fe2O7−xCa3Ti2O7(0 6 x 6 0.30)的相图及在极性和弱磁性共存区存在的磁电耦合(a)晶体结构、磁结构以及磁化强度随成分和温度的变化;(b)300 K温度下相图的局部放大图,计算铁电极化和饱和磁化强度随着成分的变化,中间区域说明极性和弱铁磁性共存;(c)在60和100 K温度下的线性磁电耦合系数随着成分的变化[31]Fig.6.Phase diagram of polycrystalline(1−x)(Sr0.4Ca0.6)1.15Tb1.85Fe2O7−xCa3Ti2O7(0 6 x 6 0.30)and the occurrence of magnetoelectric coupling in the polar and weak ferromagnetic region:(a)Dependence of crystal structure,magnetic structure and magnetization on composition and temperature;(b)cross section of the phase diagram at 300 K(saturated magnetic moment per Fe plotted with calculated polarization showing the simultaneous emergence of magnetization and polarization as x increases);(c)linear magnetoelectric susceptibility versus composition at 60 and 100 K showing that the magnetoelectric coupling increases with polarization[31]. R-P结构的杂化非本征铁电体是由位于顺电四方相布里渊区X点上的和两个非极性模耦合诱导的,其铁电极化是由A位未抵消的反铁畸变位移引起的.它不但能避免本征铁电体与磁性电子构型相斥的难题,还具有内禀的电控磁特性,有望在R-P结构中发现室温强磁电耦合的多铁性材料. R-P结构的HIF的实验研究主要集中在Ca3Ti2O7基单晶、陶瓷和薄膜以及Sr3Sn2O7陶瓷中,人们在这些材料中均确认了室温HIF的存在.相对于传统铁电材料,其剩余极化值较低,矫顽场较高.因此,如何降低矫顽场和提高剩余极化值是今后R-P结构HIF的研究重点,而许容因子的精细调控应该是解决该难题的有效手段. R-P结构的多铁性探索集中在铁基和Ca3Mn2O7基材料中.在铁基材料中,通过材料设计,获得了室温极性相与弱铁磁性相共存的多铁性材料,并在低温测得了磁电耦合效应.可惜的是,没能观察到室温非线性电滞回线,也未能测得室温磁电耦合效应.因此,获得室温非线性电滞回线和磁电耦合效应是R-P结构多铁性材料的研究机遇和挑战,而材料漏导的有效控制有望克服该难题.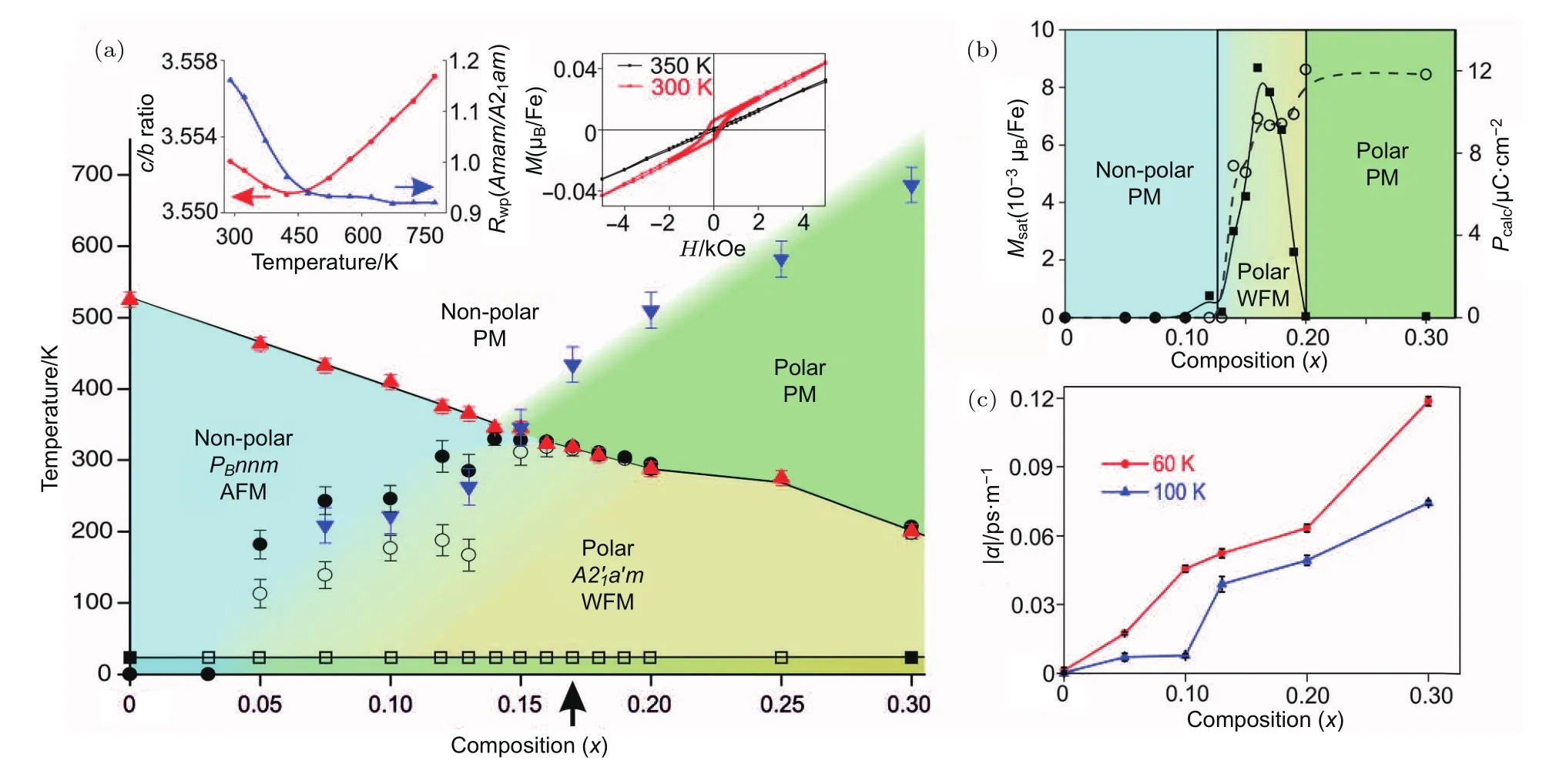
5 结论与展望
猜你喜欢
教学考试(高考化学)(2022年4期)2022-08-30
陶瓷学报(2021年3期)2021-07-22
陶瓷学报(2021年2期)2021-07-21
矿产综合利用(2020年1期)2020-07-24
新世纪智能(数学备考)(2019年9期)2019-10-16
资源再生(2019年3期)2019-04-29
资源再生(2019年1期)2019-03-04
资源再生(2019年1期)2019-03-04
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
太阳能(2015年4期)2015-02-28
