
固相萃取-超临界流体色谱-质谱联用同时快速测定中成药和保健食品中的12种抗过敏化学药物
2018-09-05 12:54:30金朦娜林丽琴
色谱 2018年9期
杨 直, 彭 彦, 金朦娜, 林丽琴
(杭州市食品药品检验研究院, 浙江 杭州 310022)
近年来,随着过敏性疾病发病率的显著提升,过敏性问题已经成为大众关注的问题[1]。目前市场上存在一些宣称有抗过敏作用的中成药和保健食品非法添加抗过敏的化学药物,特别是疗效好且起效快的产品[2]。过敏反应分4种类型,其中Ⅰ型过敏反应是最常见的类型,组胺在Ⅰ型过敏反应中占有重要作用,因此临床上应用最多的抗过敏药物就是抗组胺药[3],抗组胺药分为H1和H2受体拮抗药,与过敏反应相关的是H1受体,作为抗过敏的H1受体拮抗剂根据分子结构主要分为氨基醚类等6大类。
国内外对于抗过敏药物的检测方法主要有:高效液相色谱-紫外检测(HPLC-UV)法[2,4]、毛细管电泳(CE)法[5,6]、液相色谱-质谱联用(HPLC-MS/MS)法[7-9]、气相色谱(GC)法[10]、原子吸收光谱(AAS)法[11]等。这些检测方法均只涉及部分H1受体拮抗剂品种的检测,并没有对目前国内常用的抗过敏化学药物在中成药和保健食品非法添加领域进行系统性检测。
超临界流体色谱(SFC)分离原理与气相色谱和液相色谱一致,主要利用超临界流体兼具液体的溶解能力和气体的低黏度高扩散能力而进行物质的分离分析[12,13]。SFC适合分析热不稳定化合物、难挥发化合物、液相色谱难以分离的极性化合物[14,15]和手性化合物[16-18]。同时由于超临界流体色谱主要采用CO2作为流动相,因此还有经济、环保的优势。
超临界流体色谱作为一种成熟的分析技术还应用于中成药和保健食品的非法添加检测领域。本研究选择了12种临床上常用的H1受体拮抗剂类抗过敏化学药物进行研究,它们分别为氨基醚类(苯海拉明、茶苯海明、氯马斯汀)、乙二胺类(安他唑啉、曲吡那敏)、三环类(异丙嗪、赛庚啶、氯雷他定)、丙胺类(氯苯那敏)、哌嗪类和哌啶类(阿司咪唑、地氯雷他定、去氯羟嗪)。利用超临界流体-色谱-质谱联用技术建立了12种抗过敏化学药物的分析方法,同时考虑到中成药和保健食品基质复杂,存在基质效应,利用固相萃取的前处理技术对样品的基质干扰进行了排除,实现了复杂基质中多组分抗过敏化学药物定性、定量分析。
1 实验部分
1.1 仪器、试剂与材料
Waters ACQUITY超高效合相色谱仪(UPC2)配Xevo TQD三重四极杆质谱检测器,ACQUITY UPC2Trefoil CEL1色谱柱(150 mm×3.0 mm, 2.5 μm), Oasis MCX固相萃取柱(规格:3 mL/60 mg, 30 μm), Oasis PRiME HLB萃取柱(规格:3 mL/60 mg, 30 μm)(美国Waters公司); Milli-Q超纯水器(美国Millipore公司); KQ600DV超声波清洗器(昆山市超声仪器有限公司); METTLER XS205DU电子天平(瑞士Mettler Toledo公司)。
二氧化碳(纯度≥99.99%,上海振信公司);甲醇和乙腈(均为色谱纯,纯度≥99.9%,美国Merck公司);甲酸(色谱纯,纯度99%,美国ACS恩科化学公司);氨水(分析纯,上海凌峰化学试剂有限公司)。盐酸苯海拉明(100066-200807,纯度99.9%)、富马酸氯马斯汀(100229-200803,纯度99.9%)、盐酸安他唑啉(100216-200702,纯度100.0%)、盐酸异丙嗪(100422-201603,纯度99.5%)、盐酸赛庚啶(100502-200401,纯度100.0%)、氯雷他定(100615-201404,纯度100.0%)、马来酸氯苯那敏(100047-200606,纯度99.7%)、阿司咪唑(100301-199901,纯度100.0%)、地氯雷他定(100919-200902,纯度99.7%)、盐酸去氯羟嗪(100262-201302,纯度99.1%)、茶苯海明(100120-201504,纯度99.7%)均购自中国食品药品检定研究院;盐酸曲吡那敏(批号:108377,纯度99.5%)购自德国Dr. Ehrenstorfer公司。
1.2 标准溶液的配制
混合标准储备液:分别称取12种对照品各25 mg置于50 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为标准储备液。分别精密量取上述溶液各1 mL置于同一50 mL量瓶中,加甲醇稀释至刻度,摇匀,配制成质量浓度均为10 mg/L的混合标准储备液。
混合标准曲线溶液:用5%(体积分数,下同)氨化甲醇(含5%氨水)将混合标准储备液配制成质量浓度为5、10、20、25、50、100、200和250 μg/L的系列混合标准曲线溶液。
1.3 样品前处理
样品溶液的提取:精密称取0.2~0.3 g固体样品(片剂研细混匀,胶囊剂取内容物混匀),或精密量取2.0~3.0 mL溶液(溶液剂取样前需摇匀),置于10 mL量瓶中,加入适量甲醇,超声提取处理(40 KHz, 20 min),放置冷却至室温,用甲醇定容至刻度,摇匀,使用0.45 μm混合型滤膜过滤。精密量取续滤液1.0 mL,与9.0 mL 2%甲酸水溶液混匀,待净化。
样品溶液的净化:上述提取溶液转移至Oasis MCX萃取柱中,待提取液完全通过后,用3 mL水淋洗,抽至近干,用5 mL 5%氨化甲醇(含5%氨水)洗脱,并抽干。整个萃取过程控制流速约为1 mL/min。收集洗脱液,用0.22 μm混合型滤膜过滤,取续滤液待测。
1.4 分析条件
色谱柱:ACQUITY UPC2Trefoil CEL1色谱柱;柱温:45 ℃;流动相:A为CO2, B为0.1%氨水甲醇溶液。梯度洗脱条件:0~0.2 min, 95%A; 0.2~10.0 min, 95%A~45%A; 10.0~12.0 min, 45%A; 12.0~14.0 min, 45%A~95%A。背压:12.4×106Pa;流速:1.2 mL/min;进样量:0.5 μL。
离子源:电喷雾离子源,除茶苯海明(以8-氯茶碱为检测物质)采用负离子模式(ESI-)外,其余均采用正离子模式(ESI+);扫描方式:多反应监测(MRM);离子源温度(TEM): 150 ℃;毛细管电压:3 000 V;脱溶剂温度:500 ℃;脱溶剂气流速:1 000 L/h;锥孔反吹气流速:50 L/h。12种抗过敏化学药物的母离子、子离子、锥孔电压和碰撞能量参数详见表1。
2 结果与讨论
2.1 前处理条件的优化
本研究中的12种化学药物在甲醇中的溶解性均较好,考虑到甲醇是极性较大的有机溶剂,样品浸润能力较好,选择甲醇为样品提取溶剂。超声提取时间考察了10、20和30 min,综合考虑样品的浸润时间和提取效率,最终选择超声时间为20 min。
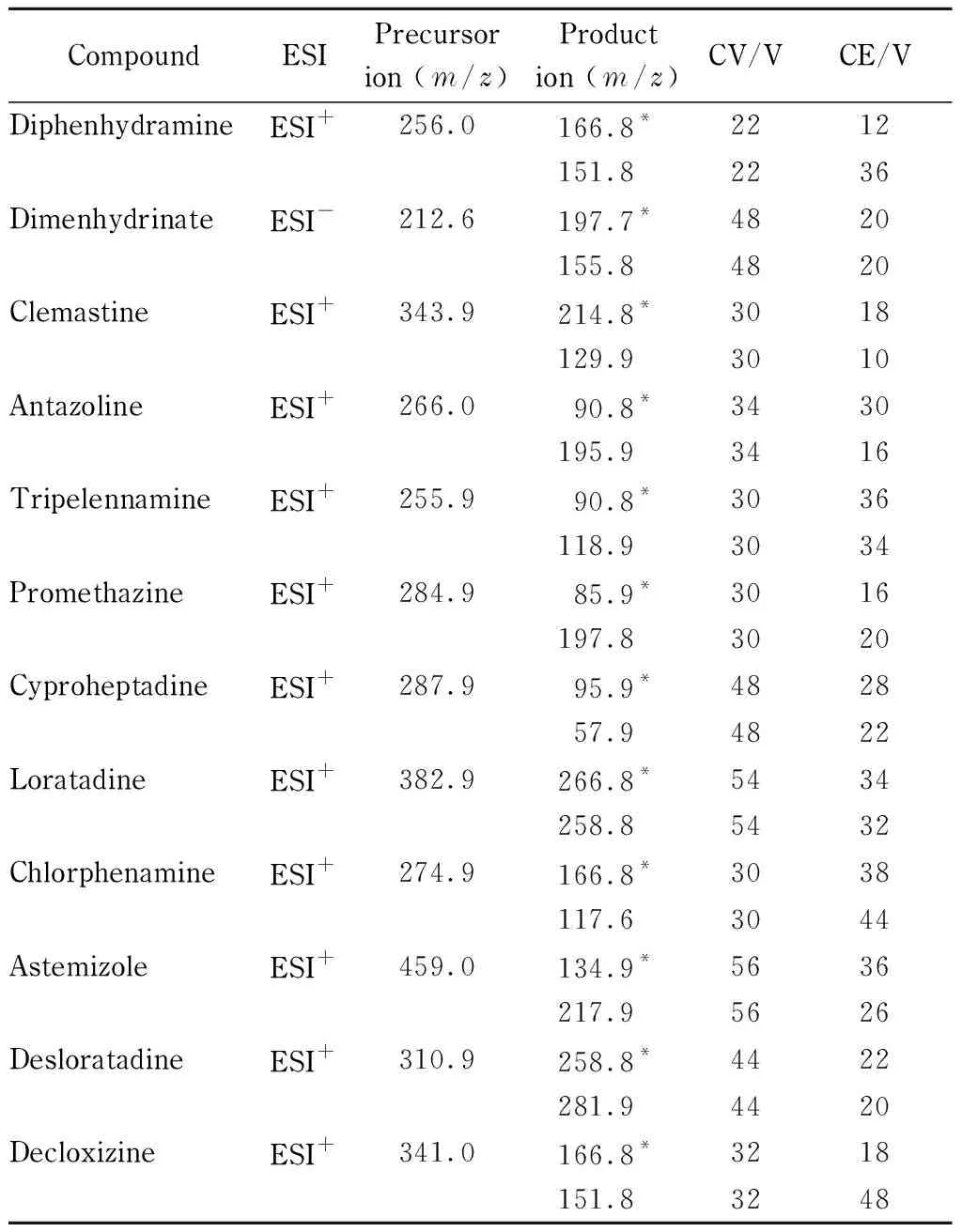
表 1 12种抗过敏化学药物的母离子、子离子、锥孔电压和
* Quantitative ion.
12种化学药物均属于有机碱类化合物,分子结构上基本都含有O、N、S等电负性的杂原子,容易获得质子,pKa值基本在7.5~9.5之间,具有弱碱性。结合化合物的结构特点,考察了具有较强反相吸附作用力的强亲水性Oasis PRiME HLB萃取柱和具有离子吸附和反相吸附混合作用力的Oasis MCX固相萃取柱对样品的萃取净化能力。采用Oasis PRiME HLB萃取柱时,12种化学药物的平均回收率为65%~118%,特别是氯雷他定,回收率仅为65%;采用Oasis MCX萃取柱时,12种化学药物的平均回收率为85%~96%,符合分析要求,这与Oasis MCX萃取柱具备两种结合能力有关。
同时还考察了样品待净化溶液的组成、淋洗液和洗脱液的种类3个影响提取效率的因素。比较了甲醇提取液与水、2%甲酸溶液、4%甲酸溶液和8%甲酸溶液以1∶10(v/v)混合制备的4种样品待净化溶液。比较了水、10%甲醇、20%甲醇和30%甲醇4种淋洗液。比较了3、5和10 mL甲醇及3、5和10 mL 5%氨化甲醇6种洗脱液。通过优化条件,最终选择用2%甲酸溶液进行甲醇提取液的酸化,3 mL水淋洗,5 mL 5%氨化甲醇洗脱为最优前处理条件。
2.2 色谱条件的优化
SFC的原理融合了气相色谱和液相色谱的特点,配合SFC专用色谱柱能达到最佳的分析效果。本研究考察了UPC2配套使用3根色谱柱(Trefoil CEL1、Trefoil AMY1和Trefoil CEL2)时待分析成分的保留时间、相邻峰的分离度以及峰拖尾情况,Trefoil CEL1各方面表现最佳。
二氧化碳为非极性流动相,要进行有机碱类化合物的分离,需要在流动相中添加极性溶剂进行改性,本研究考察了甲醇、乙醇、乙腈3种添加剂。同时为了减少峰拖尾现象,添加了0.1%氨水增加流动相的pH值,可抑制待分析化合物的电离,减少与色谱柱中硅醇基的相互作用。在考察的添加剂中,甲醇极性最大,流动相极性可调范围最大,并且与样品溶液的溶剂一致,兼容性较好,峰形对称,并且化合物的保留时间相对较短,因此选择0.1%氨水甲醇溶液为添加剂。
色谱系统的背压与流速相关联,在1.2 mL/min的流速下考察了11.0×106、12.4×106和14.5×106Pa 3个背压条件。随着背压的升高,色谱峰的保留时间提前,主要是由于背压升高导致超临界二氧化碳的密度升高,增加了流动相的洗脱能力。综合考虑各化合物的保留时间和分离度,选择背压为12.4×106Pa。
2.3 质谱条件的优化
取适量混合标准储备液,分别在正离子模式和负离子模式下进行测试。茶苯海明由8-氯茶碱和苯海拉明组成,鉴于苯海拉明属于待测化合物之一,因此以8-氯茶碱作为检测对象,间接测定茶苯海明的含量。12种化学药物中除8-氯茶碱采用ESI-模式以外,其他11种化合物均采用ESI+模式。同时在确定各化合物的MRM参数时,采用软件IntelliStart自动进行质谱方法的开发和优化,确定子离子碎片、最佳锥孔电压和碰撞电压。以曲吡那敏为例,其母离子、子离子、锥孔电压和碰撞电压的优化见图1。

图 1 曲吡那敏的母离子、子离子、锥孔电压和碰撞电压优化Fig. 1 Optimization of parent ion, daughter ion, zone voltage, and collision voltage of tripelennamine
2.4 方法学验证
2.4.1线性关系
将混合标准曲线溶液按照优化后分析条件进行测定,12种抗过敏化学药物的总离子流图见图2。以质谱多反应监测(MRM)中化合物定量子离子的峰面积(Y)为纵坐标,化合物的质量浓度(X, μg/L)为横坐标进行线性回归,结果见表2。数据表明,12种化学药物在相应的浓度范围内线性关系良好,相关系数(r)均大于0.998。
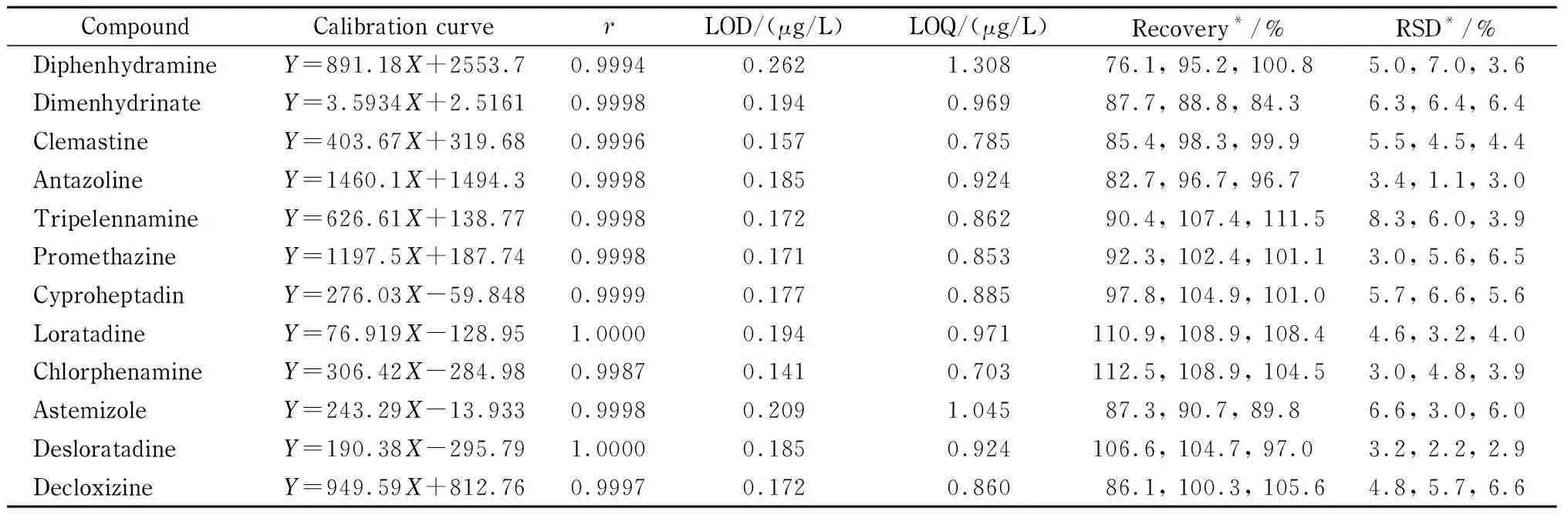
表 2 12种抗过敏化学药物的校准曲线、相关系数、检出限、定量限和回收率(n=6)
* Values from left to right represented the results of low, medium and high spiked levels;Y: peak area;X: mass concentration, μg/L.
2.4.2检出限和定量限
取空白片剂基质样品,研细混匀,精密称取相当于一片的量,置于10 mL量瓶中,加入一定量的混合标准储备液,然后按照1.3节方法处理,进行检测,以各化合物二级质谱中的定量离子为参考指标,12种化学药物的检出限及定量限检测结果见表2。
2.4.3准确度和精密度
取空白片剂基质样品,研细混匀,精密称取约相当于一片的量,置于10 mL容量瓶中,分别加入0.5、1和5 mL混合标准储备液,每个水平制备6份样品,进行加标回收率的评价。准确度(回收率)和精密度(RSD)结果见表2。
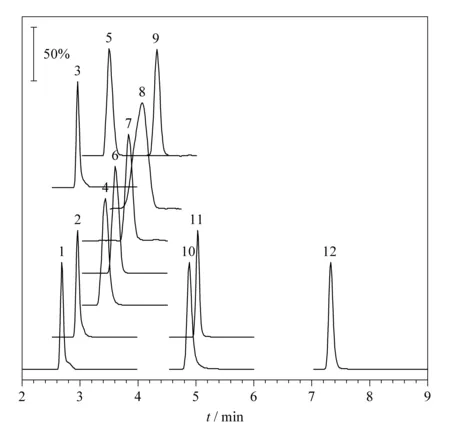
图 2 12种抗过敏化学药物的总离子流色谱图Fig. 2 Total ion chromatograms of the 12 anti- allergic chemical drugs Peak Nos.: 1. diphenhydramine; 2. chlorphenamine; 3. tripelennamine; 4. promethazine; 5. clemastine; 6. decloxizine; 7. cyproheptadine; 8. loratadine; 9. dimenhydrinate; 10. desloratadine; 11. antazoline; 12. astemizole.
2.4.4稳定性
取混合标准曲线溶液(质量浓度为100 μg/L),分别于放置0、1、3、5、8 h时后进样测定,比较各化合物的峰面积。结果表明,12种化学药物峰面积的RSD为2.9%~8.3%,表明溶液在8 h内基本稳定。
2.5 实际样品分析
应用本方法对市售的10批中成药和11批保健食品进行定量分析,样品的剂型包括片剂、胶囊剂、溶液剂和颗粒剂,基本涵盖了常见剂型。结果在6批次样品中检出2种抗过敏的化学药物,氯苯那敏的添加量为0.41~14.6 mg/g,苯海拉明的添加量为1.69 mg/g。本研究组在建立超临界流体色谱-质谱联用的分析方法同时还建立了高效液相色谱分析方法,通过对样品检测结果的比较发现,两种测定方法的结果基本一致(见表3)。

表 3 21批次样品测定结果
3 结论
本研究建立了经固相萃取净化处理,利用超临界流体色谱-质谱联用技术快速测定中成药和保健食品中12种抗过敏化学药物的分析方法,该方法稳定性较好,准确、快速,可作为筛选抗过敏化学药物非法添加的补充检验手段。但是本研究未对该12种抗过敏化学药物的衍生物进行研究,因此不能用于可能具有相同功效的衍生物的筛查工作,对于该类衍生物的分析研究有待下一步工作的开展。
猜你喜欢
云南化工(2021年5期)2021-12-21 07:41:20
食品安全导刊(2021年20期)2021-08-30 06:39:48
粉末冶金技术(2021年3期)2021-07-28 06:26:50
童话世界(2017年29期)2017-12-16 07:59:32
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
当代化工研究(2016年5期)2016-03-20 16:21:35
四川电力技术(2015年5期)2015-12-19 11:04:54
电力建设(2015年2期)2015-07-12 14:15:58
应用化工(2014年11期)2014-08-16 15:59:13
特产研究(2014年4期)2014-04-10 12:54:22
