
随访十年的脊髓小脑性共济失调3型一家系研究及伦理学分析
2018-08-31 03:26康健捷项薇邓兵梅杨红军彭凯润王伟民
卒中与神经疾病 2018年4期
康健捷 项薇 邓兵梅 杨红军 彭凯润 王伟民
脊髓小脑性共济失调( Spinocerebellar ataxias,SCAs)是一大类由于遗传因素造成的神经系统变性疾病,多为常染色体显性遗传,占神经系统遗传性疾病的10%~15%,患病率为1~4/10万[1]。SCAs有明显的遗传异质性和基因多效性,所以患者的临床表现错综复杂、病情轻重不一,对于临床分型就增加了很大的难度,随着分子生物学的发展,基因诊断已成为诊断SCAs亚型的标准,目前已有30多种[2]致病基因被定位,其中脊髓小脑性共济失调3型(SCA3)是亚洲人种最常见的亚型,多呈常染色体显性遗传。本研究结合相关文献对1个随访10年的SCA3家系发病的临床特征、影像学特点和基因型及相关伦理学进行讨论,以提高临床医生对SCA3的认识。
1 资料与方法
1.1 家系调查 先证者为女性,广东籍患者,因行走不稳5年、言语不清2年于2007年1月18日收住本院,先证者的外婆、母亲及舅舅也有类似症状,未就医,均已故,具体死因不详。遂展开家系调查。2007年该家系4代15名成员中共有5人发病。随访10年,截止2017年1月该家系4代21名成员中共有7人发病。
1.2 研究对象 家系中I2、II2、II5、Ⅲ2、Ⅲ5、Ⅲ7、Ⅲ20为患者, 共7例(其中I2、II2、II5已故,为回顾性诊断 ),另6例来自上述家系的第2代、第3代和第4代的无症状的“健康”个体:II4、Ⅲ4、Ⅲ8、Ⅲ9、Ⅲ11、Ⅳ1。
1.2 方 法
1.2.1 知情同意 按照知情同意原则,向所有申请基因诊断者说明基因测试的目的、程序、可能出现的情况、目前所能检测的范围,并保证保守秘密,尊重其隐私权,如果受试人无法理解或为18岁以下未成年人,则向其陪护人、父母做出解释。
1.2.2 基因检测 先证者为Ⅲ5,女性,2012年对先证者进行SCAl、SCA2、SCA3、SCA6、SCA7、SCAl2 和齿状核红核苍白球丘脑路易斯核萎缩(dentatorubral pallidoluysian atrophy,DRPLA) 致病基因检测,明确SCA亚型,确诊为SCA3。2013年对Ⅲ7检测相应异常基因,2015年对Ⅲ20检测相应异常基因,2016年对II4、Ⅲ4、Ⅲ8、Ⅲ9、Ⅲ11、Ⅳ1检测相应异常基因。抽取外周静脉血2 mL,依地酸钠钙(EDTA) 抗凝,采用全血基因组DNA 抽取试剂盒提取基因组DNA作为PCR扩增模板。引物设计、PCR扩增以及DNA测序由广州金域医学检验中心完成。
1.2.3 遗传咨询 检测资料通知受试者本人, 未成年人则通知其父母,向他们解释SCA遗传方式、基因突变形式、临床主要症状、目前的治疗状况和护理相关知识,详细宣教产前诊断相关知识,并鼓励患者积极配合治疗以改善生活质量。
2 结 果
2.1 家系调查 截止2017年1月该家系4代人中有7人有步态不稳等表现,其中Ⅰ2、Ⅱ2、Ⅱ5、Ⅲ5表现为行走不稳、言语不清、饮水呛咳,已死亡。Ⅰ2:42岁起病,59岁去世;Ⅱ2:39岁起病,56岁去世;Ⅱ5:30岁起病,55 岁去世;Ⅲ2:32岁起病,现40岁,吐字不清、饮水呛咳、扶手杖行走,生活自理;Ⅲ5:26岁起病,36岁窒息死亡;Ⅲ7:28岁起病,现36岁,吐字不清、饮水呛咳、行走不稳、扶手杖行走,生活自理;Ⅲ20:24岁起病,现26岁,需搀扶下才能行走,发音困难、吞咽困难。3代发病平均年龄分别为42、34.5、27.5岁。其家系见图1。
2.2 临床表现、体征、辅助检查、致病基因测序和头颅磁共振检查 患者家系成员检测SCA3相应异常基因显示:无临床症状的Ⅱ4、Ⅲ4、Ⅲ8、Ⅲ11、Ⅳ1的SCA3相关基因的CAG 重复数为14~41次,属于正常范围。无临床症状的Ⅲ9,30岁,查体可见双侧巴氏征阳性,CAG 重复数为77次,确诊为SCA3症状前患者。7例患者和1例症状前患者的临床表现、体征和致病基因检测、核磁共振成像(表1)。患者均无认知功能障碍,无感觉障碍。实验室检查:肝、肾功能、血电解质、血脂、肿瘤指标、甲状腺功能、叶酸、维生素B12 血浓度、肌酶、铜蓝蛋白、免疫学指标均正常。影像学检查:肝、胆、脾、胰、肾B超均未见异常。

图1 SCA3患者的家系图
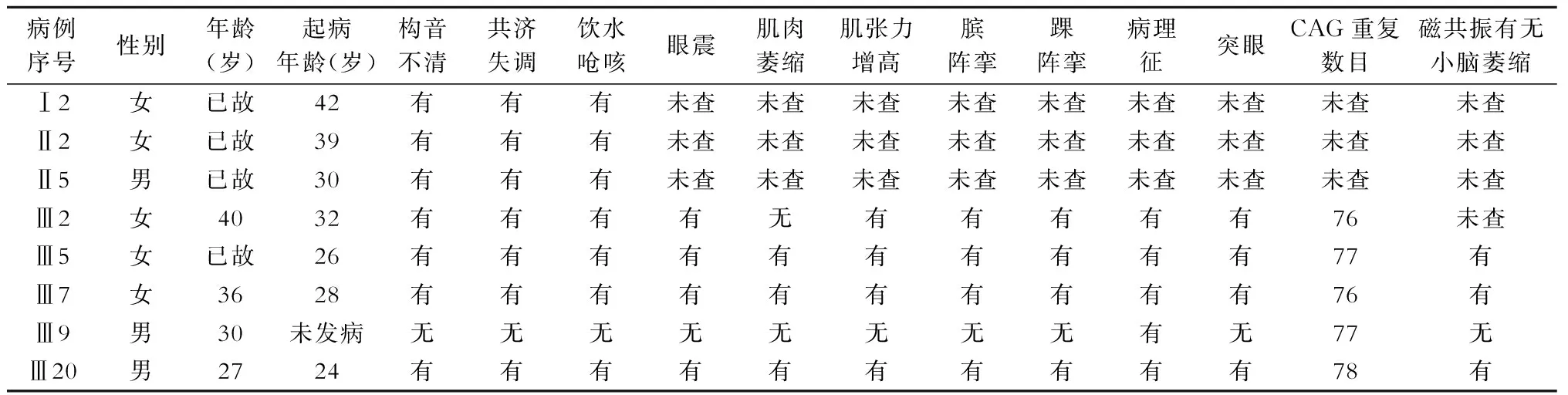
表1 7例患者和1例症状前患者发病特点及异常等位基因CAG重复数目
2.2.1 病例1(先证者) Ⅲ5,女性,26岁起病,因行走不稳5年、言语不清2年于2007年1月住院诊治。患者于2002年1月出现行走欠稳,2003年2月出现双手动作笨拙、步伐变小、步态不稳,2005年1月出现言语不清,饮水呛咳、吞咽困难、全身消瘦明显、四肢肌肉萎缩,行走时蹒跚步态,容易跌倒,需扶手杖行走,2007年1月发音困难,需家人搀扶下缓慢行走,长期卧床,生活不能自理,2012年8月因窒息死亡。2007年1月查体:体型消瘦,对答切题,构音欠清,语速缓慢,小脑性语言。定向力、记忆力、计算力正常。眼球上视受限,其余各向活动正常,瞳孔等大、等圆,直径约3.0mm,对光反射灵敏,双眼水平眼震,闭目有力,鼻唇沟对称,伸舌居中,可见舌肌颤搐,悬雍垂居中,双侧咽反射迟钝。四肢肌肉萎缩,肌张力稍增高,肌力Ⅴ-级,四肢腱反射亢进,双侧霍夫曼征(+),双侧踝阵挛(+),双侧巴氏征(+)。深浅感觉对称正常。双手意向性震颤,双侧指鼻试验、跟膝胫试验不稳不准,轮替动作笨拙,双上肢反击征(+)。不能独自站稳,搀扶下行走呈宽基底步态,闭目难立征(+)。颈软,无抵抗。头颅MRI检查提示小脑和脑干萎缩(图2)。2012年2月查体:不能站立行走,鼻饲饮食,重度营养不良,发音困难,突眼,四肢肌肉萎缩,肌张力增高,其余查体同前。基因检测:进行SCAl、SCA2、SCA3、SCA6、SCA7、SCAl2 和DRPLA致病基因检测和DNA测序显示,SCA3 相关基因的CAG 重复数为77 次(图3)(正常参考值<44次),属于全突变范围,符合SCA3 的基因突变特征,其他亚型SCAl、SCA2、SCA6、SCA7、SCAl2、DRPLA相关基因的CAG 重复数属于正常范围,确诊为SCA3。
2.2.2 病例2 Ⅲ2,女性,40岁。因行走不稳8年、言语不清5年于2016年12月就诊。患者于2008年2月出现行走欠稳,步基变宽,2011年4月出现言语欠清,讲话不流畅,有饮水呛咳,现经口进食,尚可独自行走,行走不稳。基因检测显示SCA3 相关基因CAG 重复数为76次。

图2 先证者Ⅲ5的头颅MRI显示小脑和脑干萎缩

图3 先证者Ⅲ5的基因检测显示异常基因片段CAG重复数目为77次
2.2.3 病例3 Ⅲ7,女性,36岁。因行走不稳4年、言语不清1年于2013年11月就诊。患者于2009年5月出现行走欠稳,步基变宽,2012年7月出现言语欠清,讲话不流畅,偶有饮水呛咳。头颅MRI检查提示小脑萎缩。2016年3月行走不稳明显,尚可独自行走,经口进食,有饮水呛咳,语言不流利。2013年基因检测显示SCA3 相关基因CAG 重复数为76次。
2.2.4 病例4 Ⅲ20,男性,27岁。因行走不稳2年、言语不清1年于2015年7月就诊。患者于2013年6月出现行走欠稳,症状进展较快,2014年7月出现言语欠清,讲话不流畅,饮水呛咳,行走不稳加重,容易跌倒,需要扶手杖行走,2016年6月搀扶下行走,进食困难,发音困难。头颅MRI检查提示小脑萎缩。2015年7月基因检测显示SCA3 相关基因CAG 重复数为78次。
2.2.5 症状前患者 Ⅲ9,男性,30岁。无行走欠稳等临床症状。2016年12月行基因检测显示SCA3 相关基因CAG 重复数为77次。
2.3 治 疗
给予Ⅲ5、Ⅲ7、Ⅲ20巴氯芬、加巴喷丁减轻肢体肌肉痉挛和疼痛。给予Ⅲ7丙戊酸钠2年余,临床症状逐渐缓慢加重,按时监测肝肾功能、血常规、血氨。Ⅲ20曾在外院进行干细胞移植治疗,无明显疗效,
3 讨 论
SCA3是一种延迟的神经系统常染色体显性遗传性退行性疾病,具有高度的临床异质性,它是脊髓小脑性共济失调中最为常见的亚型,几乎占所有SCA患者的50%[3-4];病变主要累及小脑, 也可影响到桥脑、延髓、橄榄、脊髓和视神经;发病年龄20~60岁,但大多于20~40岁起病[5-6]。临床表现以小脑性共济失调和构音障碍为突出表现,SCAs 各亚型间临床表现较为相似,仅依照其临床症状进行分型及确诊非常困难。因为SCAs 各亚型致病基因及突变位点不同,所以通过基因突变检测可以将其区别开来[7]。
该家系7例患者均表现为行走不稳、构音不清、饮水呛咳,神经系统阳性体征为小脑性共济失调、延髓麻痹和锥体束征,3例患者头颅MRI检查提示小脑萎缩,定位于小脑和延髓。患者均隐匿起病,逐渐进展,结合患者有明确的家族史,3代人中均有患者,男、女均发病,定性诊断首先考虑为常染色体显性遗传的神经系统变性病:遗传性小脑性共济失调。对先证者Ⅲ5进行SCA3 相关基因的CAG 重复数为77次,属于全突变范围,符合SCA3 的基因突变特征,其他亚型SCAl、SCA2、SCA6、SCA7、SCAl2、DRPLA相关基因的CAG重复数属于正常范围,确诊为SCA3。文献有报道少数SCA3 家系患者可出现认知功能障碍[8-11]与嗅觉障碍[12]、听力障碍[13],表明SCA3 具有很高的表型异质性。本家系患者未出现上述症状。
2007年该家系4代15名成员中共有5人发病,其中3人已死亡。随访10年,先证者Ⅲ5于发病10年窒息死亡,Ⅲ2、Ⅲ7、Ⅲ20分别于2008年、2009年、2013年发病,截止2017年1月该家系4代21名成员中共有7人发病。除第4代由于年龄尚小,在2个月~13岁之间未发病, 其余每代均有患者。双亲中有一方为患者,子代必有该病患者(Ⅲ2的子代已经经过基因检测CAG重复数属于正常范围,Ⅲ7、Ⅲ9的子代年龄尚小,均未发病)。双亲正常者其子代无发病, 如II4和Ⅲ4的SCA3相关基因的CAG重复数属于正常范围,其子代也不会发病,符合常染色体显性遗传。该家系前3代24~42岁发病,即早期一般不发病,达到一定年龄才表现出明显的临床症状,如在随访中Ⅲ2、Ⅲ7、Ⅲ20发病年龄分别为32、28、24岁。第4代由于年龄较小,均未发病。3代发病平均年龄分别为42、34.5、27.5岁。发病年龄逐代提前,病情发展速度逐代加快,符合遗传早现现象。
脊髓小脑性共济失调3型又称马查多约瑟夫病(Machado-Joseph disease,MJD)。因SCA3和MJD的疾病基因位于相同位点即14 q32 12而合称为脊髓小脑性共济失调3型/马查多约瑟夫病(SCA3/MJD)。研究显示SCA3/MJD导致编码蛋白ataxin-3羧基端形成异常扩展的多聚谷氨酰胺(polyglutamine,PolyQ)肽链被认为是引起病理改变的蛋白质基础,从而引起蛋白的错误折叠[14],在神经系统中形成泛素阳性核内包涵体(neuronal intranuclear inclusions,NIIs),通过蛋白质-蛋白质相互作用产生选择性神经细胞毒性作用[15]。正常人CAG重复次数为12~42次,患者为52~84 次,且其CAG异常重复次数越多,发病越早且越重[16-19]。本家系中CAG 重复数最多的为Ⅲ20是78次,发病年龄24岁,发病较早,行走不稳进展较快,发病3年已不能独自行走,有进食困难、发音困难,病情较重。本家系Ⅲ5的CAG重复数为77次,发病年龄26岁,发病5年已经长期卧床,生活不能自理,发病10年因窒息去世。而Ⅲ2、Ⅲ7的CAG重复数均为76次,她们均在行走欠稳发病3年出现言语不清,并进行性加重,但发病年龄稍晚,分别为32和28岁,病情较轻,发病8年尚可独自行走,可经口进食,这种现象可能是由于Ⅲ5的CAG 重复数多于后两者,这与报道的发病规律一致。但Ⅲ9的CAG重复数为77次,与Ⅲ5相同,但目前30岁,尚未发病,这种现象可能与性别等遗传异质性有关。由于不同患者具有不同的遗传基础,从而导致发病年龄、病程进展、病情严重程度以及预后等都可能不同。
目前,SCA3尚无特异性治疗,主要是对症治疗,巴氯芬、加巴喷丁减轻肢体肌肉痉挛和疼痛,丙戊酸钠[20]、伐伦克林[21]、拉莫三嗪[22]可能改善临床症状、延缓病情的进展。目前体外试验和动物实验基因治疗显示在细胞培养实验中利用RNA干扰技术有选择地让SCA3的致病基因产生了“沉默”,而不影响正常基因的功能[23],但用于临床还很遥远。近年来,立体定向神经干细胞移植手术治疗小脑萎缩已在临床上开展,针对SCAs 患者的治疗已经取得了一定疗效,这可能为SCAs 患者的治疗带来新的希望[23-25]。不过,本家系Ⅲ20曾在外院进行干细胞移植治疗无明显疗效。正因为SCA3尚无特异性治疗,所以面对患有SCA3的患者,医生的职责不仅在于提高临床诊断和基因检测水平,更要通过长期随访,及时调整对症治疗用药,提高治疗效果以改善患者的生活质量,同时建立患者对医生的信任并增加对此疾病的认识,促进家族成员积极地进行基因检测,筛选症状前患者,并提供遗传咨询、产前诊断以防止带有异常基因的胎儿出生,这有利于提高患者的优生优育。而现在所面临的问题是虽然基因诊断较传统诊断方法能更早更准确地诊断疾病及分型, 但同时可加重患者的心理负担, 若患者的隐私不能得到保护, 将会带来一系列的社会歧视与舆论压力,严重损害了患者的婚姻生活和就业等权益,所以患有遗传病的家系成员一般都是出于此顾虑,才迟迟不愿接受基因检测。但因SCA3属于延迟的神经系统遗传病,大多于20~40岁起病,许多患者在生育时尚未发病,也未行基因检测,不知自己是否会患病及遗传给后代,从而生活在对此病的疑虑和恐惧的阴影之中,严重影响家庭生活和工作。因此,医生对整个家系的心理疏导非常重要,详细的遗传咨询和疾病相关知识的宣教对每个家系成员都意义重大。该家系中Ⅲ4经过基因检测已经明确未遗传SCA3异常基因,确定了自己和孩子均不会发病,才终于卸下心理包袱,可以轻松健康的生活。家系成员Ⅲ9尚无临床症状,但不知自己将来是否会发病,在随访医生的耐心宣教下接受了基因检测,虽然经基因检测CAG重复数目为77次,被确诊为SCA3症状前患者,致病基因的外显率为100%,但是当再次生育下一代时可进行产前诊断以防止带有异常基因的胎儿出生,保证孕育健康的后代。家系成员Ⅲ11因为有一侧肢体麻木乏力,怀疑自己也遗传了此病而情绪低落,尤其是其妻子怀孕以后担心胎儿也可能被遗传这种疾病,所以计划人工流产并且提出离婚。经过随访医生仔细问诊和查体,诊断患者的肢体麻木乏力是由颈椎病所致,基因检测结果也证实CAG重复数目在正常范围内,不会发病,避免了人工流产和离婚的结局。总之,长期随访和全程的心理疏导能够帮助患者更好地建立信心去工作和生活以及积极配合治疗,促进家族成员更积极地进行基因检测,让症状前患者及其家人明白此病易受外显率的影响而病情轻重不一,不要产生恐惧心理,经过产前基因诊断可以避免家族缺陷延续。同时,应该制定更加完善的法律法规,确保个人隐私不受侵犯,避免遭受歧视,并加强相关知识的宣教,保障患者心理健康。
综上所述,SCA3为一组神经系统遗传性疾病,临床特征以小脑性共济失调和构音障碍为突出表现,具有高度异质性,CAG 重复序列数目的检测对于基因诊断和症状前诊断是一种十分有效的方法。临床要严格按照医学伦理规范进行严谨的遗传检测。长期有效的随访和心理辅导可促进家族成员进行基因筛查,有助于发现症状前患者及促进产前诊断,对患病家系的优生优育意义重大。
猜你喜欢
中国医药导报(2022年28期)2022-11-25
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
好孩子画报(2019年8期)2019-09-19
创新作文(小学版)(2019年4期)2019-07-24
郑州大学学报(医学版)(2019年3期)2019-06-03
中南林业科技大学学报(2019年4期)2019-04-08
