
肺鳞癌并亚急性运动神经元病1例并文献复习
2018-08-27 06:16:12王潇李青云李昕昱来青伟胡朋樊红彬
神经损伤与功能重建 2018年8期
王潇,李青云,李昕昱,来青伟,胡朋,樊红彬
亚急性运动神经元病(subacute motor neuronopathy,SMN)是神经副肿瘤综合征的一种罕见特殊类型,发病率<2%[1],临床上误诊率较高。SMN主要侵及脊髓前角细胞及延髓运动神经核。SMN的原发肿瘤以淋巴瘤和骨髓瘤多见,尤以淋巴瘤多见[2]。该病多表现为亚急性进行性进展,主要为上、下运动神经元受损,以下运动神经元损害多见。脑脊液检查蛋白的含量可正常或增高,细胞数基本正常,肌电图表现为失神经电位[3]。病理变化表现为大量脊髓前角细胞的脱失和退行性变,脊髓白质可见片状的脱髓鞘改变。SMN发病机制尚不明确[3]。目前尚无统一诊断标准,尚无特效治疗。现报道1例肺癌合并SMN的案例。
1 临床资料
患者,男性,75岁,慢性起病,2017年4月因“言语不清、四肢无力1年,加重2月”入院。既往高血压病史8年余,糖尿病史1年余,腰椎间盘突出摘除术后9年余(植入钢板)。患者8年前于外院确诊肺鳞癌,行右肺大部切除术,术后化疗1周,患者感不适拒绝进一步化疗,后反复复查胸部CT平扫+增强未见明显复发及转移灶。吸烟史20年余,1包/d。1年前患者无明显诱因下出现言语不清、左下肢无力,1周左右后逐渐感右下肢无力,后逐渐累及上肢,均以近端为著,有舌肌萎缩,无明显四肢肌肉萎缩,偶有饮水呛咳、吞咽困难,外院诊断“脑梗死”。出院后长期服用抗血小板、稳定斑块类药物,但仍感症状进行性加重。2月前患者感言语不清、饮水呛咳加重,且有口角流涎、明显四肢无力感,双下肢为著,同时逐渐出现右手大鱼际肌、指间肌萎缩。病程中患者无头晕、头痛,无恶心、呕吐,无意识障碍,无腹痛腹泻,精神饮食睡眠一般,大小便正常自控。
入院查体:体温36.5℃,脉搏78次/分,呼吸18 次/分,血压150/70 mmHg(1 mmHg=0.133 kPa);全身皮肤粘膜无黄染,未见出血点,全身浅表淋巴结未触及到明显肿大;双肺听诊呼吸音清,未及干湿啰音;心律齐,心脏各瓣膜听诊区未及病理性杂音;腹平软,无压痛及反跳痛,无异常包块;双下肢未见水肿。神经系统查体:神志清楚,言语不清,双侧瞳孔等大等圆,约3 mm,光敏,眼动灵活充分,无眼震,无复视,双侧鼻唇沟对称,伸舌居中,可见舌肌萎缩,舌肌纤颤。双上肢肌力5-级,左下肢肌力4-级,右下肢肌力4+级,右手大鱼际肌、指间肌萎缩,伴肌颤。四肢肌张力正常,四肢腱反射减弱。指鼻试验稳准,疲劳试验阴性。深浅感觉正常,右侧巴氏征(+),左侧巴氏征可疑阳性。辅助检查:空腹血糖6.22 mmol/L。男性肿瘤全套:胃泌素释放肽前体[Pro-GRP]95.25 pg/mL。其余生化、血常规、病毒全套、甲功、糖化血红蛋白、凝血功能等血液学检查未见明显异常。胸部CT示:“右肺癌术后”改变;两肺间质性改变。头颅平扫+增强CT示:多发性脑梗死(双侧半卵圆中心、基底核区及侧脑室旁);皮质下动脉硬化性脑病;脑萎缩。肌电图示:四肢肌、面肌、舌肌见大量自发及束颤电位,并见巨大电位,募集单纯相;上下肢运动、感觉传导速度稍减慢,复合肌肉动作电位(compound muscle action potential,CMAP)波幅降低;双侧面神经潜伏期、波幅均正常。F波上下肢神经、面神经测定潜伏期、引出率基本正常。重复电刺激未见波幅变化。符合脊髓前角细胞病变,周围神经病变,见表1、2。颈胸椎MRI、心电图、心脏彩超、消化系彩超等未见明显异常。
诊断:患者有四肢无力,右手大鱼际肌、指间肌萎缩伴肌颤,四肢腱反射减弱等颈、腰段下运动神经元受累为主的临床表现,有言语不清、饮水呛咳、舌肌萎缩伴纤颤等延髓神经核受累等球部下运动神经元损害表现,同时有右侧巴氏征阳性、左侧巴氏征可疑阳性的锥体束损害表现。基本定位在前角/前根、周围神经和(或)锥体束损害。
鉴别诊断:①无肌痛、肌酶未见明显异常、肌电图检查未见肌源性损害,排除多发性肌炎、皮肌炎等肌肉疾病。②疲劳试验阴性结合肌电图重复电刺激未见波幅变化,排除重症肌无力、Lambert-Eaton等神经肌接头疾病。③无明显感觉异常,肌电图有束颤波,不支持慢性炎症性脱髓鞘性多发性神经病、慢性格林巴利等周围神经病变;肌电图有上下肢感觉传导速度稍减慢、波幅降低,可能同时合并周围神经损害。④无感觉异常及小便失禁等症状,颈胸椎MRI未见明显异常,排除脊髓病变;⑤复查头颅CT平扫+增强较前未见明显改变,排除同时合并脑梗死、出血、颅内转移等中枢性损害;⑥无外伤、感染、中毒、代谢障碍及相关家族遗传病等病史。

表1 肌电图检查结果
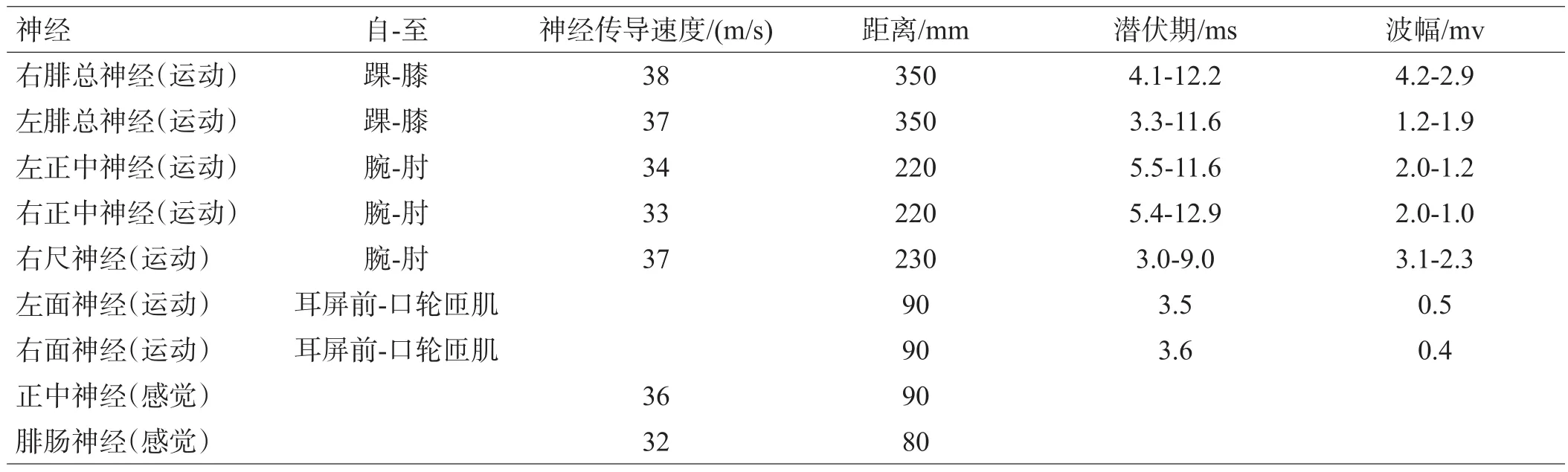
表2 神经传导速度测定结果
综上所述,最终定位在脊髓前角,同时可能合并周围神经、锥体束损害。定性诊断主要考虑为运动神经元病,同时可能合并周围神经损害。考虑该患者发病年龄较大,且有明确的肺癌病史,病情进展相对较快,不太符合经典的运动神经元病,且以单一病变不好解释,因此考虑副肿瘤性运动神经元病可能性大。副肿瘤运动神经元病的分类有:肌萎缩侧索硬化(最常见)、原发性侧索硬化、进行性肌萎缩、进行性延髓麻痹、SMN及其他类型。原发性侧索硬化一般无肌萎缩及肌肉束颤,进行性肌萎缩首发症状常常为肌肉萎缩,发病年龄小、进展慢,而进行性延髓麻痹多仅以延髓神经核受累为表现。前三者均易于区别,而肌萎缩侧索硬化在临床表现上与SMN很相似,且有报道称极少数患者SMN可同时合并肌萎缩侧索硬化,因此主要注意鉴别。肌萎缩侧索硬化多表现为上、下运动神经元同时受累,常伴有四肢腱反射亢进,双侧病理反射阳性,双下肢肌张力增高和迷走神经损害等症状,延髓麻痹多在后期出现。尤其是痉挛状态和腱反射亢进更具有鉴别意义。而该患者仅表现为以下运动神经损害为主,双下肢腱反射减弱,无明确的双侧病理征阳性,且患者的锥体束损害亦不能排除与颅内梗死灶的关系。同时,患者发病在肿瘤基本处于稳定期,病程呈亚急性起病且呈进行性发展。故最终考虑SMN。此外,患者肌电图有上下肢感觉传导速度稍减慢、波幅降低,这些特征提示有周围神经髓鞘及轴索损害,需进一步鉴别:首先,患者肌电图检查见广泛神经源性损害累及四肢肌、面肌及舌肌,且出现巨大电位,支持运动神经元病。而周围神经病变一般仅累及四肢肌,且无巨大电位出现。其次,临床表现上周围神经损害可能出现无力、肌肉萎缩和束颤,但肌无力多以远端为主,且常常伴有感觉异常。该患者既往糖尿病史容易合并周围神经损害且经历化疗后亦可能损伤周围神经,因此患者同时可能合并有周围神经病变,但周围神经病变并非本次发病的主因。
建议患者完善神经元抗体谱检测(抗HU抗体、抗Yo抗体、抗Ri抗体、抗PNMA2、抗CV2抗体、抗Amphiphysin抗体),结果均为阴性,但由于仅能在50%患者的血清中发现特异性神经元抗体或神经抗体谱不全面,故不能用于排除该诊断。SMN无特效治疗,住院期间予以改善循环、营养神经等对症治疗,患者症状较入院时未见明显变化要求出院。4月后电话随访,家属述患者言语不清较前进一步加重,饮水呛咳、吞咽困难较明显,肢体无力、舌肌萎缩、右手大鱼际肌、指间肌萎缩较前未见明显变化。
2 讨论
SMN是较为罕见的特殊类型神经副肿瘤综合征。神经副肿瘤综合征是潜在的恶性肿瘤或恶性肿瘤的远隔效应所引发的神经系统受损,而没有癌肿直接侵犯到神经组织或由于代谢性、感染性及血管性并发症的原因所致,神经副肿瘤综合征可累及中枢神经系统,周围神经系统,神经肌肉接头和肌肉本身[5]。SMN主要表现为亚急性进行性上、下运动神经元受损,尤以下运动神经元损害多见,上肢及颅神经受损较少,感觉障碍不存在或较轻微。上运动神经损害的表现类似于肌萎缩侧索硬化。SMN病程进展较缓慢,其发病通常发生在恶性肿瘤的缓解期且症状相对稳定时,与原有肿瘤的病情进展大多不一致,SMN的病程进展要相对稳定,部分患者经过数月或数年后神经症状可趋于稳定或有所改善。
SMN患者脑脊液检查蛋白含量可正常或增高,细胞数基本正常,即轻度的蛋白-细胞分离[5]。肌电图检查表现为失神经电位,运动、感觉传导速度基本上正常,F波延迟正常,重复刺激无运动后减量[3]。病理变化表现为大量脊髓前角细胞脱失和退行性变,尤其是颈段;脊髓白质可见片状的脱髓鞘改变,神经胶质纤维增粗、增多,主要见于神经元丢失最为明显的区域,未见巨噬细胞及淋巴细胞浸润。骨骼肌可见神经源性肌萎缩[4]。
SMN发病机制尚不明确,大多数学者认为与免疫功能障碍有关。猜想神经元和肿瘤细胞可能有共同的抗原决定簇,肿瘤细胞作为始动抗原导致机体产生高特异性的抗体[6],与SMN相关的高度特异性抗体主要为抗HU抗体[7],抗HU抗体在补体参与下不仅抑制肿瘤细胞的生长,也导致宿主神经系统的损伤。宿主神经系统神经元被破坏时释放的抗原进一步刺激神经系统中的B细胞导致鞘内抗体的合成,引起更强烈并且广泛的免疫应答反应[4]。另一学说认为,发病原因可能为肿瘤患者应用免疫抑制剂后造成的病毒机会性感染,病变上酷似“脊髓灰质炎”,但目前尚未分离出病毒[3,8]。
SMN尚无统一的诊断标准,主要依赖于临床表现、确诊的肿瘤证据及肌电图检查、神经元抗体检测、肌肉活检等依据。神经元抗体谱的检测对于神经副肿瘤综合征的早期发现与诊断及其相关肿瘤意义重大。血清中高滴度的神经元抗体如:抗HU抗体、抗Yo抗体、抗Ri抗体、抗Tr抗体、抗Ta抗体、抗CAR抗体、抗VGCC抗体、抗CV2抗体、抗Amphiphysin抗体等被认为是神经副肿瘤综合征的标记[9],已有部分报道发现SMN特征性的抗HU或CV2/CRMP5抗体[7]。但据统计,仅50%的患者血清中发现特异性神经元抗体[9],在SMN中仅5%患者可检测出抗Hu抗体[11],因此血清中特异性神经元抗体的缺乏亦不能作为排除诊断的标准。
目前SMN尚无特效治疗,主要是针对原发病灶的治疗,部分使用免疫治疗,但疗效不佳。该病预后不佳,但也有少数良性进展的病例。影响预后的因素较多,如年龄>60岁、神经系统多处受累和没有对原发灶及时采取治疗等,都会使死亡率增高[5]。
因此,当临床上出现原因不明的,呈亚急性进行性发展的,临床上表现为下运动神经元损害为主,尤其是合并肿瘤病史的患者,要高度怀疑SMN,同时注意与其他疾病相鉴别,积极检测神经元抗体对于诊断有重要的价值[12]。更明确的诊断离不开对SMN患者的长期随访。
