
Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease
2018-08-20 02:10:16CherilTapiaRojasNibaldoInestrosa
中国神经再生研究(英文版) 2018年10期
Cheril Tapia-Rojas, Nibaldo C. Inestrosa,
1 Centro de Envejecimiento y Regeneración (CARE UC), Departamento de Biología Celular y Molecular, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
2 Centre for Healthy Brain Ageing, School of Psychiatry, Faculty of Medicine, University of New South Wales, Sydney, Australia
3 Centro de Excelencia en Biomedicina de Magallanes (CEBIMA), Universidad de Magallanes, Punta Arenas, Chile
Funding: This work was supported by grants PFB (Basal Financing Program) 12/2007 from the Basal Centre for Excellence in Science and Technology and FONDECYT, No. 1120156 (to NCI) and a pre-doctoral fellowship from the National Commission of Science and Technology of Chile (CONICYT) (to CTR).
Abstract Alzheimer’s disease (AD) is the most common form of dementia in the older population, however, the precise cause of the disease is unknown. The neuropathology is characterized by the presence of aggregates formed by amyloid-β (Aβ) peptide and phosphorylated tau; which is accompanied by progressive impairment of memory. Diverse signaling pathways are linked to AD, and among these the Wnt signaling pathway is becoming increasingly relevant, since it plays essential roles in the adult brain. Initially, Wnt signaling activation was proposed as a neuroprotective mechanism against Aβ toxicity. Later, it was reported that it participates in tau phosphorylation and processes of learning and memory. Interestingly, in the last years we demonstrated that Wnt signaling is fundamental in amyloid precursor protein (APP) processing and that Wnt dysfunction results in Aβ production and aggregation in vitro. Recent in vivo studies reported that loss of canonical Wnt signaling exacerbates amyloid deposition in a transgenic (Tg) mouse model of AD. Finally, we showed that inhibition of Wnt signaling in a Tg mouse previously at the appearance of AD signs, resulted in memory loss, tau phosphorylation and Aβ formation and aggregation; indicating that Wnt dysfunction accelerated the onset of AD. More importantly, Wnt signaling loss promoted cognitive impairment, tau phosphorylation and Aβ1–42 production in the hippocampus of wild-type (WT) mice, contributing to the development of an Alzheimer’s-like neurophatology. Therefore, in this review we highlight the importance of Wnt/β-catenin signaling dysfunction in the onset of AD and propose that the loss of canonical Wnt signaling is a triggering factor of AD.
Key Words: Wnt signaling; Wnt target genes; Wnt/β-catenin; Alzheimer disease; amyloid-β; tau phosphorylation;memory loss; synaptic dysfunction
Alzheimer’s Disease (AD) and Wnt Signaling
AD is the most prevalent dementia, increasing drastically with age (Mayeux and Stern, 2012). It is manifested by a progressive loss of learning and memory; as a result of the accumulation of senile plaques (SPs) and neurofibrillary tangles (NFTs) in the hippocampus and cerebral cortex (Selkoe and Hardy, 2016).The most studied hypotheses to explain the onset and development of AD is the amyloid hypothesis (Selkoe and Hardy,2016). According to this cascade, amyloidogenic amyloid precursor protein (APP) processing lead to amyloid-β (Aβ) eptide formation; deregulating kinases, phosphatases, and several signaling pathways that promote tau phosphorylation, synaptic failure and neurodegeneration (Selkoe and Hardy, 2016).However, although the neuropathological signs, and potential protective mechanisms of AD have been described, the precise cause remains unclear. Interestingly, an important signaling pathway that is strongly related to AD is Wnt signaling (De Ferrari et al., 2014; Inestrosa and Varela-Nallar, 2014). This pathway is fundamental in the development of the Nervous Central System; but also plays important roles in the adult brain,regulating processes of synaptic plasticity and memory (Inestrosa and Varela-Nallar, 2014). Diverse reports have indicated that Wnt signaling is impaired in different AD mouse models(Scali et al., 2006; Toledo and Inestrosa, 2010). More important,familial AD patients with PS1 mutations have reduced β-catenin levels, strongly suggesting Wnt signaling loss (Zhang et al., 1998). More important, an allele of low-density lipoprotein receptor-related protein-6 (LRP6) associated with a loss of function of Wnt signaling is a susceptibility gene for late-onset AD (De Ferrari et al., 2007). In fact, a recent study showed that AD patients have a loss of Wnt activity, such as indicated by decreased β-catenin levels and increased tyrosine-216 phosphorylation of glycogen synthase kinase 3 (GSK-3β) compared with age-matched controls in the prefrontal cortical lobe structures of AD brains (Folke et al., 2018). Therefore, is possible that Wnt signaling activity plays a key role in the development of AD.
Wnt ligand binds to the Frizzled (Fz) receptor and LRP5/6 co-receptor, disassembling the destruction complex and inhibiting GSK-3β (Gao and Chen, 2010). This permits β-catenin to accumulate and translocate to the nucleus, where it binds to transcriptional factors T-cell factor/lymphoid enhancer factor(TCF/LEF) to induce the transcription of Wnt target genes(Nusse and Clevers, 2017). In contrast, in absence of Wnt proteins, GSK-3β becomes active, phosphorylating β-catenin and promoting its degradation via proteasome (Nusse and Clevers,2017). The Wnt signaling family is composed of 19 ligands and 10 Fz receptors, which depending on the cellular context and ligand-receptor binding could activate Wnt/β-catenin dependent or independent signaling (Mikels and Nusse, 2006;Grumolato et al., 2010). In the neurons, the Wnt-Fz complexes are expressed spatially and temporally distinct in the cell (Varela-Nallar et al., 2012). Fz1 is located at the presynaptic region,promoting synaptic differentiation in hippocampal neurons(Varela-Nallar et al., 2009); Fz2 is mainly located at the neuronal soma (Varela-Nallar et al., 2012); Fz3 is expressed in dendrites(Varela-Nallar et al., 2012), regulating axonal outgrowth, guidance, and neural tube closure (Wang et al., 2002; Lyuksyutova et al., 2003); Fz5 control the survival of mature neurons in the thalamus (Liu et al., 2008), and it is localized in growth cones during development of hippocampal neurons (Varela-Nallar et al., 2012; Slater et al., 2013), where regulates synaptogenesis in the hippocampus (Sahores et al., 2010); Fz7 expression decreases during hippocampal development with a distribution in the soma and neurites (Zhao et al., 2005) and Fz9 is important for hippocampal development, due its enrichment at the growth cone (Varela-Nallar et al., 2012) and it is expressed along the processes of the post-synaptic region in mature hippocampal neurons (Ramirez et al., 2016).
Wnt signaling has important functions in the adult brain.Among these, it regulates the presynaptic structure (Inestrosa and Varela-Nallar, 2014), inducing the clustering of synapsin I(Lucas and Salinas, 1997; Hall et al., 2000) and the α7-nicotinic acetylcholine receptor (nAChR) (Farias et al., 2007); promoting the endocytosis and recycling of vesicles and increasing the pre-synaptic sites (Cerpa et al., 2008). Also, stimulated dendritogenesis (Ciani et al., 2011), induced neurotransmitter release(Cerpa et al., 2008), facilitates the induction of LTP (Chen et al., 2006) and its activity is modulated in spatial memory tasks(Tabatadze et al., 2012), playing a role in synaptic plasticity and cognition. The majority of the canonical Wnt pathway effects are mediated by the transcription of Wnt target genes; however several effects also could be occurring by a mechanism that did not required changes in the expression of Wnt target genes ref-jnc. An example is Wnt-7a in hippocampal neurons, which induces the clustering of presynaptic proteins without altering the expression of genes in response to β-catenin (Cerpa et al.,2008). But, independent of the mechanism, it is essential to highlight that synaptic failure is an early event in the AD pathology (Terry, 1991; Selkoe, 2002), supporting the possibility that Wnt signaling activity could be the key in AD pathogenesis.
Role of Wnt Signaling in the Pathogenesis of AD
Almost twenty years ago, our laboratory first proposed a relation between the Wnt signaling pathway and AD, suggesting that activation of Wnt/β-catenin signaling is protective against amyloid toxicity in vitro (De Ferrari and Inestrosa, 2000). Later, diverse reports have validated the neuroprotective role of the Wnt signaling pathway against the damage induced by Aβ species, preventing tau phosphorylation and preserving the cognitive abilities in vivo using mouse models of AD (Toledo and Inestrosa, 2010; Vargas et al., 2014; Rivera et al., 2016).We showed that activation of Wnt signaling by ANDRO (Andrographolide) (Tapia-Rojas et al., 2015a) ameliorates spatial memory loss, reduces the activation of inflammatory processes,and decreases the levels of Aβ species and tau phosphorylation in the hippocampus of a natural model of AD (Rivera et al.,2016). Similarly, in the triple transgenic (3×) transgenic (Tg)-AD mouse, activation of Wnt pathway reduces tau phosphorylation, Aβ formation, and prevents apoptosis (Jin et al., 2017).More importantly, our laboratory recently demonstrated that ANDRO improves learning and memory, recovery synaptic transmission, and reduces the levels of phosphorylated tau and Aβ aggregates in aged Octodon degus, a natural model of AD(Rivera et al., 2016). Thus, the protective role of Wnt signaling observed in vivo propose that activation of Wnt signaling pathways could be used as a potential treatment to impede the progression of AD.
Interestingly, studies have also suggested that the loss of Wnt signaling could be involved in the etiology of both familial AD(FAD) and sporadic AD (SAD). Concerning to FAD, mutations in the γ-secretase component Presenilin 1 (PS1) blocks the Wnt/β-catenin pathway (Zhang et al., 1998); promoting GSK-3β activity, tau hyper phosphorylation (Takashima et al.,1998), β-catenin degradation (Zhang et al., 1998) and repressing the transcriptional activities of the transcriptional co-factors of TCF/β-catenin (Francis et al., 2006). Also, conditional Lrp6 gene deletion in a mouse model of AD stimulates amyloid pathology (Liu et al., 2014), suggesting that Wnt signaling dysfunction could be involved in the early appearance and severity in genetic cases of AD (Baki et al., 2004). In SAD, the role of Wnt signaling in the initiation of AD is supported by studies that indicate that a polymorphism in the GSK-3 promoter increases its activity and is a risk factor for late-onset AD (Mateo et al.,2006). Also, an allele of the LRP6 co-receptor related to Wnt signaling dysfunction is a susceptibility gene for SAD (De Ferrari et al., 2007). Finally, chronic lithium medication reduces the prevalence of AD in patients with bipolar disorders (Forlenza et al., 2012), suggesting that activation of Wnt signaling might prevent processes necessary for the initiation and progression of SAD.
We recently demonstrated that loss of Wnt signaling activity could be a factor triggering the onset and progression of AD. In our last study, we were the first to demonstrate that inhibition of the Wnt signaling in the J20 Tg mouse model of AD, previous to the appearance of AD signs, accelerates the onset and development of the pathology, including accelerated Aβ production and aggregation, tau phosphorylation, and memory loss(Tapia-Rojas and Inestrosa, 2018). Interestingly, in the same study we reported that dysfunction of Wnt signaling triggered a neurodegenerative AD-like process in wild-type (WT) mice,leading to Aβ formation, tau phosphorylation, and alterations in learning and memory (Tapia-Rojas and Inestrosa, 2018). Thus,the loss of Wnt signaling could be considered a potential cause of AD and activation of Wnt signaling could prevent the development of the pathology.
In the following sections, we will describe studies that demonstrate the influence of canonical Wnt activity on the production and aggregation of Aβ, tau phosphorylation and synaptic failure that leads to memory loss, the main events associated to the AD neuropathology.
Wnt signaling promotes Aβ production
The Aβ peptide is produced by amyloidogenic APP processing,in which the APP protein is cleaved by the β-secretase enzyme and subsequently by the γ-secretase complex to generate products of 39–43 amino acids that are highly toxic to neurons (De Strooper et al., 2010). Amyloidogenic APP processing is abnormal and the causes that lead to this process in the brain are unidentified (Masters and Selkoe, 2012). Several studies have suggested that Wnt signaling could play a role in Aβ production. A Wnt agonist of the Wnt pathway in N2a cells decreases the Aβ peptide concentration and represses the transcription of the BACE (β-secretase enzyme β-site APP-cleaving enzyme)through the binding of β-catenin to TCF4 (Parr et al., 2015).In contrast, loss of Wnt function using a β-catenin shRNA significantly increases BACE1 mRNA and its protein expression(Parr et al., 2015). Similarly, LRP6 knockdown favors the formation of APP fragments via the amyloidogenic pathway, such as C-terminal fragment (CTF)β and the Aβ peptide (Liu et al.,2014).
Interestingly, we have demonstrated that inhibition of the Wnt/β-catenin pathway increases the Aβ1–42concentration and the Aβ42/Aβ40 ratio, favoring the formation of Aβ oligomers in vitro (Tapia-Rojas et al., 2016). Specifically, our results show that blocking canonical Wnt signaling at different scales results in amyloidogenic proteolytic processing of APP, increasing the levels of C99, Aβ1–42peptide, Aβ42/Aβ40ratio, and low-molecular-weight Aβ oligomers such as trimers and tetramers(Tapia-Rojas et al., 2016). In contrast, we showed that activation of the Wnt pathway favors non-amyloidogenic APP processing,reducing Aβ1–42peptide formation and aggregation, with the concomitant increment in Aβ1–40peptide (Tapia-Rojas et al.,2016). The ability of canonical Wnt signaling to impede Aβ aggregation was also reported previously in our laboratory, using WASP-1 (Wnt-activating small molecule potentiator-1), a synergic molecule of the Wnt ligand Wnt-3a that enhances activation of the pathway, which blocks Aβ aggregation in in vitro assays(Vargas et al., 2015). More importantly, using mutant forms of β-catenin that are incapable of binding to the TCF/LEF transcription factor (β-cat ΔTCF), we showed for the first time that activation of canonical Wnt signaling requires of the transcription of Wnt target genes to switch APP processing to the non-amyloidogenic APP pathway, indicated by the formation of C83 fragment, and prevents Aβ1–42production and formation of oligomers favoring the Aβ1–40peptide generation (Tapia-Rojas et al., 2016). Finally, we proposed that Wnt signaling regulates APP processing, possibly by a mechanism that involves changes in the expression of BACE1 enzyme, because overexpression of β-catenin activate Wnt target genes and reduce BACE1 protein levels, in contrast to β-cat ΔTCF (Tapia-Rojas et al., 2016). This mechanism is relevant, because BACE1 inhibition also reduces Aβ in plasma, cerebrospinal fluid (CSF), and brain samples of rats, monkeys, and in AD patients (Kennedy et al., 2016). Therefore, our results indicate that inhibition of the Wnt/β-catenin signaling pathway promotes amyloidogenic APP processing, Aβ1–42formation and aggregation.
It was previously reported that deletion of the Lrp6 gene in an APP-PS1 mouse model of AD aggravates amyloid deposition in the brain of these animals (Liu et al., 2014), suggesting that Wnt signaling also plays a role in the amyloid pathology in vivo. In addition, our laboratory demonstrated that activation of Wnt signaling in aged Octodon degus reduced Aβ1–40and Aβ1–42peptides, decreased the levels of Aβ oligomers, and the number of senile plaques in the hippocampus (Rivera et al., 2016), indicating that Wnt signaling activation prevents the formation of the Aβ peptide and the development of amyloid deposits. Interestingly and in contrast to the previously mentioned, we recently demonstrated that Wnt signaling dysfunction accelerates Aβ1–42production in the hippocampus of a J20 Tg AD mouse model and promotes Aβ1–42formation in the hippocampus of WT mice (Tapia-Rojas and Inestrosa,2018). The increased concentrations of Aβ1–42peptide in the hippocampus of these animals was accompanied by an increase in the Aβ42/Aβ40ratio, suggesting higher toxicity in the brain of both Tg and WT animals only by inhibiting Wnt/β-catenin signaling (Tapia-Rojas and Inestrosa, 2018). This effect is similar to that previously reported in FAD patients, in which an increased Aβ42/Aβ40ratio has been directly related with severe forms of the pathology (Hellstrom-Lindahl et al., 2009). We also reported that in treated mice, the levels of Aβ1–42in the CSF were decreased (Tapia-Rojas and Inestrosa, 2018), suggesting altered clearance of Aβ possibly as consequence of Aβ aggregation. In fact, we observed a major presence of Aβ soluble species in the brain of animals exposed to Wnt inhibitors and in J20 Tg mice we detected the presence of Aβ species that could correspond to hexamers and do-decamer soluble aggregates (Tapia-Rojas and Inestrosa, 2018), which are considered synaptotoxic (Lesné et al., 2006, 2013). The latter is consistent with reports that have indicated that few alterations in the Aβ42/Aβ40ratio promote Aβ aggregation forming oligomers (Yoshiike et al., 2003; Kuperstein et al., 2010). Additionally, in Tg mice with deficient function of Wnt signaling we observed increased number of senile plaques (Tapia-Rojas and Inestrosa, 2018), validating the in vitro studies that indicated that Wnt signaling loss lead to Aβ production and aggregation.
Importantly, we reveal that Wnt signaling inhibition trigger the formation Aβ1–42and increase the Aβ42/Aβ40ratio in the hippocampus of WT mice, effect that was accompanied of a reduction in Aβ1–42concentration in the CSF. The Aβ pathology observed in WT mice is consistent with previous studies,in which we demonstrated that a high methionine diet induces dysfunction of canonical Wnt signaling at the same time that it promotes Aβ formation, oxidative stress and memory impairment (Tapia-Rojas et al., 2015b). Altogether, our in vitro and in vivo studies demonstrate that inhibition of Wnt/β-catenin signaling promotes amyloidogenic APP processing, Aβ1–42production, increased Aβ42/Aβ40ratio and formation of aggregates;which are fundamental for promoting toxicity in the AD pathology (McLean et al., 1999).
The amyloid accumulation is promoted by an imbalance between Aβ production and clearance. Microglia in the brain plays an important role in Aβ clearance by a variety of phagocytic and digestive mechanisms. Among these mechanisms, TREM2(an innate immune receptor and a Type I transmembrane protein) regulates phagocytosis and promotes microglial survival,contributing to Aβ elimination (Ries and Sastre, 2016). Interestingly, TREM2 promotes microglial survival by activating the Wnt/β-catenin signaling pathway, suggesting that Wnt pathway is necessary to the correct clearance of amyloid deposits (Zheng et al., 2017). Another possibility to degrade Aβ is promoting the expression of Insulin degrading enzyme (IDE). Unpublished data of our laboratory suggest that IDE is a target gene of the Wnt pathway, therefore Wnt signaling also could modulates Aβ degradation. More studies are necessary to validate these possibilities.
Wnt signaling regulates tau phosphorylation
Tau phosphorylation is mediated by diverse protein kinases which can phosphorylate tau at different sites (Mandelkow and Mandelkow, 2012). Among these enzymes, GSK-3β phosphorylates practically all tau residues described in AD (Kremer et al., 2011; Hernandez et al., 2013). GSK-3β is a central component of Wnt signaling and its activity is mediated by the function of the Wnt/β-catenin pathway (Metcalfe and Bienz,2011). GSK-3β inhibition impedes the formation of NFTs observed in a double transgenic of GSK-3β and tau protein(Engel et al., 2006). Our laboratory showed that Wnt signaling activation by ANDRO reduces tau phosphorylation in young and old APP-PS1 AD mouse model (Serrano et al., 2014). A similar effect was observed in aged Octodon degus treated with ANDRO, where the levels of phosphorylated Thr231,Ser235, and both Ser202 and Thr205 (AT8 epitope) were decreased (Rivera et al., 2016). Synergic activation of canonical Wnt signaling by WASP-1 in APP-PS1 mice also reduced PHF-1 (Ser396 and Ser404) tau phosphorylation (Vargas et al., 2015), strongly suggesting that Wnt activation prevent tau phosphorylation.
In contrast, mutations in PS1 promote GSK-3β activity and tau phosphorylation (Takashima et al., 1998). Conditional transgenic mice overexpressing GSK-3β in the adult brain showed decreased β-catenin levels in a nuclear fraction, increased tau phosphorylation, and neuronal death (Lucas et al., 2001). Inhibition of Wnt signaling favors GSK-3β-mediated tau hyper phosphorylation (Scali et al., 2006; Hooper et al., 2008). Local infusion of Dkk1, a canonical Wnt antagonist, leads to astrocyte activation, PHF1 tau phosphorylation, and neuronal death in the hippocampus of rats, effect that was blocked by lithium administration (Scali et al., 2006). In our last study we reported that attenuation of canonical Wnt signaling in J20 Tg mice accelerated the appearance of phosphorylated tau epitopes, including Thr231,Ser235, and AT8 (Tapia-Rojas et al., 2016). Surprisingly, we also have reported the presence of phosphorylated tau residues in WT mice blocking Wnt pathway. Inhibition of Wnt signaling by a high methionine diet also lead to increased phosphorylation at Ser235 and Thr231 sites in the hippocampus of WT mice (Tapia-Rojas et al., 2015b). This same effect was observed using specific inhibitors of the canonical Wnt signaling pathway, which increased phosphorylation at Ser235,Thr231, PHF1 and AT8 epitopes in the hippocampus of WT mice. Then, inhibition of Wnt signaling activates kinases that can phosphorylate tau, specifically to GSK-3β, and thus could favor its dissociation of the microtubules.
Memory loss mediated by canonical Wnt signaling dysfunction
Memory loss is the final consequence of AD pathology and according to the neurodegeration progress, memory impairment is more evident (Shankar and Walsh, 2009). The amyloid hypothesis proposes that loss of cognitive abilities is product of the synaptic failure mediated by soluble Aβ species (Selkoe and Hardy, 2016). Wnt signaling plays key roles regulating synaptic structure and function (Speese and Budnik, 2007; Oliva et al.,2013). Additionally, Wnt signaling activation occur in spatial learning (Tabatadze et al., 2012) and result in the transcription of Wnt target genes important to memory consolidation, such as calmodulin-dependent protein kinase type IV (CaMKIV)(Arrazola et al., 2009). Thus, activation of Wnt/β-catenin signaling recovers the hippocampus-dependent cognitive impairment in an APP-PS1 mouse model of AD (Toledo and Inestrosa, 2010; Serrano et al., 2014) and in Octodon degus (Rivera et al., 2016). The precise mechanism involved in the restoration of memory mediated by Wnt signaling activation in AD models is yet unknown, but possible mechanism could include the cholinergic system, because activation of canonical Wnt pathway promotes the pre-synaptic localization of α7-nAChRs (Farias et al.,2007) and the canonical ligand Wnt3a induce the differentiation of adipose-derived stem cells into cholinergic neurons, were the expression of Wnt3a increases and correlates with an increased expression choline acetyltransferase (Liu et al., 2012).
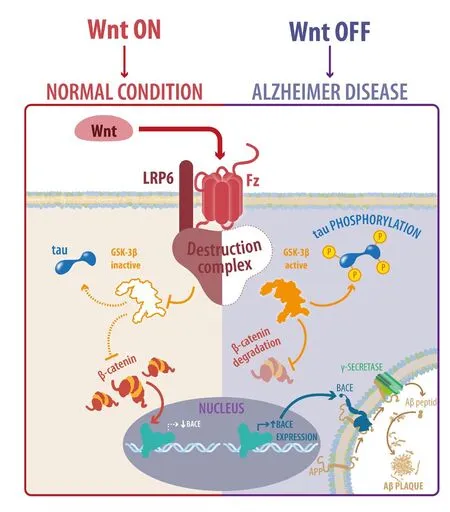
Figure 1 Wnt signaling activity is crucial in the pathogenesis of Alzheimer’s disease (AD).
Interestingly, an increase in the levels of the Wnt/β-catenin antagonist Dkk-1 is necessary to produce Aβ-mediated synaptic loss(Purro et al., 2012), suggesting that Wnt signaling has a central role in synaptic failure and memory loss Aβ-induced. Thus, Wnt signaling loss accelerates the appearance of cognitive impairment in J20 Tg mouse model of AD. More important, Wnt signaling impairment leads to altered long-term potentiation (LTP) in WT mice, a neurophysiological correlate of learning and memory(Chen et al., 2006), and triggered severe defects in spatial learning and memory in WT mice. Only blocking canonical Wnt pathway is possible to promote spatial memory loss (Tapia-Rojas and Inestrosa, 2018) and impaired recognition memory in WT mice(Fortress et al., 2013), suggesting that Wnt signaling activity modulates process of learning and memory by a mechanism independent of Aβ. Therefore, canonical Wnt signaling dysfunction is a key factor inducing learning and memory impairment and could be relevant in neurodegenerative diseases such as AD.
Conclusions
It is now known that the canonical Wnt signaling pathway is not only a protective agent in AD. Wnt/β-catenin signaling activity is fundamental for the onset of AD (Figure 1); signaling inhibition can accelerate the appearance and development of AD neuropathology and memory loss in a Tg mouse model of AD. More importantly, Wnt signaling dysfunction is sufficient to promote a neuropathological process that involves the development of three AD hallmarks: i) production and aggregation of Aβ, ii) tau phosphorylation and iii) hippocampus-dependent cognitive impairment. Thus, loss of the canonical Wnt pathway could be considered a triggering factor of the AD pathogenesis and the use of activators of the Wnt pathway could be used to prevent the development of AD-like dementia. Currently, there are no public clinical trials testing the beneficial effects of Wnt activation in a therapeutical treatment of AD. The challenge is to prevent the loss of the function of the Wnt pathway during aging, in order to prevent the appearance and development of AD.
Author contributions: Study concept, design, drafting the manuscript:CTR and NCI. Both authors read and approved the final manuscript.
Conflicts of interest: The authors declare that they have no conflicts of interest.
Financial support: This work was supported by grants PFB 12/2007 from the Basal Centre for Excellence in Science and Technology and FONDECYT, No. 1120156 (to NCI) and a pre-doctoral fellowship from the National Commission of Science and Technology of Chile (CONICYT)(to CTR).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers: Yun-Bae Kim, Chungbuk National University,Korea; Paulina Carriba, Cardiff University, UK.
Additional file: Open peer review reports 1, 2.
- 中国神经再生研究(英文版)的其它文章
- Validity and reliability of the Ocular Motor Nerve Palsy Scale
- Mitogen-activated protein kinase phosphatase 1 protects PC12 cells from amyloid beta-induced neurotoxicity
- High-frequency (50 Hz) electroacupuncture ameliorates cognitive impairment in rats with amyloid beta 1–42-induced Alzheimer’s disease
- Kaempferol attenuates cognitive deficit via regulating oxidative stress and neuroinflammation in an ovariectomized rat model of sporadic dementia
- Combined VEGF/PDGF improves olfactory regeneration after unilateral bulbectomy in mice
- Comparison of morphological and functional outcomes of mouse sciatic nerve repair with three biodegradable polymer conduits containing poly(lactic acid)