
Mitogen-activated protein kinase phosphatase 1 protects PC12 cells from amyloid beta-induced neurotoxicity
2018-08-20 02:10:28YueGuLianJunMaXiaoXueBaiJingJieXiuFangZhangDongChenXiaoPingLi
中国神经再生研究(英文版) 2018年10期
Yue Gu , Lian-Jun Ma , Xiao-Xue Bai, Jing Jie Xiu-Fang Zhang Dong Chen Xiao-Ping Li
1 Department of Respiratory Medicine, the First Hospital of Jilin University, Changchun, Jilin Province, China
2 Endoscopy Center, the China-Japan Hospital of Jilin University, Changchun, Jilin Province, China
3 Cadre’s Wards, the First Hospital of Jilin University, Changchun, Jilin Province, China
4 Department of Pediatrics, the First Hospital of Jilin University, Changchun, Jilin Province, China
Abstract The mitogen-activated protein kinase (MAPK) signaling pathway plays an important role in the regulation of cell growth, proliferation, differentiation,transformation and death. Mitogen-activated protein kinase phosphatase 1 (MKP1) has an inhibitory effect on the p38MAPK and JNK pathways,but it is unknown whether it plays a role in Aβ-induced oxidative stress and neuronal inflammation. In this study, PC12 cells were infected with MKP1 shRNA, MKP1 lentivirus or control lentivirus for 12 hours, and then treated with 0.1, 1, 10 or 100 μM amyloid beta 42 (Aβ42). The cell survival rate was measured using the cell counting kit-8 assay. MKP1, tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) mRNA expression levels were analyzed using quantitative real time-polymerase chain reaction. MKP1 and phospho-c-Jun N-terminal kinase (JNK)expression levels were assessed using western blot assay. Reactive oxygen species (ROS) levels were detected using 2′,7′-dichlorofluorescein diacetate. Mitochondrial membrane potential was measured using flow cytometry. Superoxide dismutase activity and malondialdehyde levels were evaluated using the colorimetric method. Lactate dehydrogenase activity was measured using a microplate reader. Caspase-3 expression levels were assessed by enzyme-linked immunosorbent assay. Apoptosis was evaluated using the terminal deoxynucleotidyl transferase dUTP nick end labeling method. MKP1 overexpression inhibited Aβ-induced JNK phosphorylation and the increase in ROS levels. It also suppressed the Aβ-induced increase in TNF-α and IL-1β levels as well as apoptosis in PC12 cells. In contrast, MKP1 knockdown by RNA interference aggravated Aβ-induced oxidative stress, inflammation and cell damage in PC12 cells. Furthermore, the JNK-specific inhibitor SP600125 abolished this effect of MKP1 knockdown on Aβ-induced neurotoxicity. Collectively, these results show that MKP1 mitigates Aβ-induced apoptosis, oxidative stress and neuroinflammation by inhibiting the JNK signaling pathway, thereby playing a neuroprotective role.
Key Words: nerve regeneration; mitogen-activated protein kinase phosphatase 1; c-Jun N-terminal kinase signaling pathway; Alzheimer’s disease; neurons; dementia; apoptosis; RNA interference; lentivirus; inflammation; oxidative stress; neural regeneration
Introduction
The economic and social burden of Alzheimer’s disease(AD) is an important public health problem, particularly as the prevalence of AD is increasing along with the aging population. The pathological features of AD include neuronal loss,intracellular neurofibrillary tangles and extracellular amyloid beta (Aβ) protein deposition (Goedert, 2015; Amit et al., 2017;Liu et al., 2017). Aβ plays a pivotal role in AD, and aggregated Aβ is observed in brain tissue in the early stage of AD (Puzzo et al., 2015; Golde, 2016; Müller et al., 2017; VanItallie, 2017).Aβ induces neuronal apoptosis, neuroinflammation and oxidative stress by regulating multiple signaling pathways, including mitogen-activated protein kinase (MAPK), protein kinase C and phosphoinositide 3-kinase/Akt (Smith et al., 2006; Petersen et al., 2007; Shafi, 2016; Ridler, 2017). However, the molecular mechanisms by which Aβ regulates these signaling pathways remain largely unknown. Therefore, studies on the complex signaling networks that are altered by Aβ are urgently needed to elucidate the pathogenesis of AD and aid in the development of effective clinical treatments for the disease.
The MAPK signaling pathway plays a critical role in the regulation of cell growth, proliferation, differentiation, transformation and death (Liu et al., 2012; Li et al., 2014; Thouverey and Caverzasio, 2015; Zhou et al., 2015; Yao et al., 2017). In mammalian cells, there are four MAPK signal transduction pathways, including the c-Jun N-terminal kinase (JNK, also known as stress-activated MAPK), p38 kinase (p38 MAPK),extracellular signal-regulated protein kinase (ERK) and large mitogen-activated protein kinase (BMK1/ERK5) pathways(Tiedje et al., 2014; Zhou et al., 2015; Lanna et al., 2017; Latrasse et al., 2017). Numerous studies have shown that the MAPK signaling pathway plays a major role in the pathophysiology of AD (Giraldo et al., 2014; Petrov et al., 2016; Lee and Kim, 2017). McDonald et al. (1998) found that Aβ activates the p38 MAPK pathway in rat glial cells. p38 MAPK activation in the hippocampus and cortical region is associated with neurofibrillary tangles, senile plaques, vacuolar degeneration and dystrophic neurites in post-mortem AD brain tissue (Zhu et al.,2000; Johnson and Bailey, 2003). Moreover, Aβ42injection into the basal ganglia in rats for 7 days leads to significant activation of p38 MAPK, accompanied by the activation of glial cells and neuroinflammation (Giovannini et al., 2002), whereas inhibition of the p38 MAPK pathway alleviates Aβ-induced neurotoxicity(Xu et al., 2014). Similarly, JNK signaling is activated by Aβ and is involved in Aβ-induced human neuronal damage (Troy et al.,2001; Tare et al., 2011). In addition, it has been reported that JNK activation results in excessive Aβ deposition, which in turn activates a positive feedback loop that accelerates the progression of AD (Zhu et al., 2002). These findings indicate that the p38 MAPK and JNK signaling pathways play important roles in the pathogenesis of AD. However, the molecular mechanisms underlying these processes are largely unknown.
MAPK phosphatase 1 (MKP1) is a member of the MAPK phosphatase family and is the main negative regulator of p38 MAPK, JNK and ERK (Chi and Flavell, 2008; Lawan et al.,2013; Low and Zhang, 2016). MKP1 is involved in oxidative stress, inflammatory reactions and apoptosis, and it plays a key role in the occurrence and progression of many human diseases by regulating the MAPK signaling pathway (Wancket et al., 2012; Ma et al., 2015; Moosavi et al., 2017). However, it is unknown whether MKP1 plays a role in Aβ-induced oxidative stress and inflammation in neurons.
In this study, we investigated the role of MKP1 in Aβ-induced neurotoxicity. To this end, we assessed the production of reactive oxygen species (ROS), expression of tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), and apoptosis in Aβ-treated PC12 cells. Furthermore, we examined the effect of pharmacological inhibition of JNK and the effects of MPK1 overexpression and siRNA-mediated silencing.
Materials and Methods
Cell culture
PC12 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum in a 5% CO2incubator at 37°C. The experiments were performed in accordance with the “Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals”, published by the International Committee of Medical Journal Editors (http://www.icmje.org/recommendations/).
Aβ42 preparation and cell treatment
Aβ42peptide (Sigma-Aldrich, St. Louis, MO, USA) was incubated in Tris/HCl (50 mM, pH 7.4) at a concentration of 1 mg/mL at room temperature for a minimum of 2 days for aggregation. For the experiments, Aβ42solution was diluted and added to culture media at different concentrations (0.1, 1, 10 or 100 μM).
Lentivirus infection
PC12 cells were infected with MKP1 short hairpin RNA (shRNA), MKP1 lentiviral particles or control lentivirus (Hanheng,Shanghai, China) at a multiplicity of infection of 10 in 5 mg/mL Polybrene (Sigma-Aldrich) for 12 hours and treated with 10 µg/mL puromycin (Santa Cruz Biotechnology, Dallas, TX,USA) for 21 days, according to the manufacturer’s protocol.
Cell counting kit-8 assay
Cells were plated onto 96-well plates (4000 cells/well) the day before treatment. After treatment with different concentrations (0.1, 1, 10 or 100 μM) of Aβ, 10 μL of cell counting kit-8 reagent (Sigma-Aldrich) was added to each well and cultured for 2 hours. The absorbance at 450 nm was read on a microplate reader (Bio-Rad, Hercules, CA, USA).
Quantitative real time-polymerase chain reaction(qRT-PCR)
Total RNA was extracted with Trizol (Invitrogen, Carlsbad,CA, USA), and cDNA synthesis was performed using the Superscript III Reverse Transcriptase kit (Invitrogen). The relative levels of target gene mRNA to control GAPDH mRNA were determined by qRT-PCR using TransStartTM SYBR Green qPCR Supermix (Transgene, Beijing, China) on the ABI
7500 PCR instrument (ABI, Foster City, CA, USA). The data were analyzed with the 2-ΔΔCtmethod. The primer sequences are listed in Table 1.
Western blot assay
Total cellular protein was extracted with RIPA lysis buffer (Cell Signaling Technology, Boston, MA, USA), and the concentration of total protein was determined by the BCA method(Song et al., 2017). Total protein, 15 μg, was separated by 10%sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene fluoride membranes.Membranes were blocked with 5% bovine serum albumin(Boster, Wuhan, China) at room temperature for 2 hours and immunoblotted overnight at 4°C with primary rabbit anti-rat polyclonal antibodies (1:1000; Abcam) against MKP1, p-JNK(JNK1 + JNK2 + JNK3) and GAPDH, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit polyclonal secondary antibodies (1:4000; Abcam). After extensive washing, protein bands were visualized by an ECL plus chemiluminescence kit (Beyotime Institute of Biotechnology,Haimen, China). Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD,USA).
2′ ,7′ -Dichlorofluorescein diacetate (DCFH-DA) detection of ROS
To measure intracellular ROS levels, 2 × 105PC12 cells were seeded into 6-well plates. Following treatment, the cells were treated with 20 nM DCFH-DA (Sigma-Aldrich) for 30 minutes. The green fluorescence signal was observed and photographed under a fluorescence microscope (Olympus, Tokyo,Japan).
Detection of mitochondrial membrane potential
Following treatment, the cells were incubated with 10 μg/mL JC-1 (Sigma-Aldrich) at 4°C in the dark for 30 minutes. After washing with PBS, the cells were subjected to flow cytom-

Table 1 Primer sequences
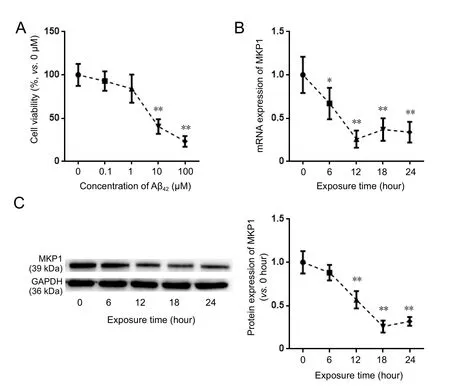
Figure 1 Eあect of Aβ42 on MKP1 expression in PC12 cells.
MKP1: Mitogen-activated protein kinase phosphatase 1; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1 beta; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.etry with an excitation wavelength of 520 nm and an emission wavelength of 596 nm. The median fluorescence intensity was recorded by the flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Assessment of superoxide dismutase (SOD) activity and malondialdehyde (MDA) levels
Intracellular SOD activity and MDA levels were measured using SOD and MDA assay kits (Jiancheng, Nanjing, China)in accordance with the manufacturer’s instructions. In brief,after cell lysis and centrifugation at 12,000 × g for 10 minutes, the supernatant was added to test reagents and incubated at 95°C in a water bath for 40 minutes. After cooling for 10 minutes, the reaction mixture was centrifuged at 4000 × g for 10 minutes, and the OD of the supernatant was measured at a wavelength of 532 nm. The SOD and MDA levels were calculated according to the OD values.
Detection of lactate dehydrogenase (LDH) activity
To assess damage caused by Aβ to cells, LDH activity was measured with the LDH detection kit (Jiancheng) as previously described (Song et al., 2016). Briefly, after Aβ treatment, 20 μL of the cell culture supernatant was mixed thoroughly with 25 μL of detection buffer and 5 μL of coenzyme I, and incubated in a water bath at 37° C for 15 minutes. Thereafter, 25 μL of 2′ ,4′ -dinitrophenylhydrazine was added to the mixture and incubated at 37°C for 15 minutes. Then, 5 minutes after the addition of 250 μL of 0.4 M NaOH, the absorbance was measured at 450 nm with a microplate reader.
Enzyme-linked immunosorbent assay (ELISA)
Cells were added to 500 μL of cold carbonate buffer (100 mM Na2CO3, 50 mM NaCl with pH 11.5) with protease inhibitors and homogenized by sonication. The cell lysate was centrifuged at 12,000 × g for 45 minutes and the supernatant was used for determination of caspase-3 content with the Caspase-3 ELISA kit (Abcam, Hong Kong, China) using a microplate reader.
Terminal deoxynucleotidyl transferase dUTP nick end labeling(TUNEL) staining
Cells were fixed with 4% paraformaldehyde at room temperature(25°C) for 20 minutes. After washing three times with PBS,the cells were permeabilized with 1% Triton X-100 and blocked with 3% H2O2at room temperature for 10 minutes. After three washes with PBS, TdT enzyme reaction solution (Coolrun,Shenzheng, China) containing TRITC-5-dUTP and TdT enzyme was added to the cells and incubated in the dark at 37°C for 60 minutes. Following nuclear staining with 5 μg/mL DAPI, the cells were observed by fluorescence microscopy.
Statistical analysis
SPSS 17.0 for Windows (SPSS, Chicago, IL, USA) was used for data processing. The data are expressed as the mean ±SD. Intergroup comparison was performed using analysis of variance (with α = 0.05).
Results
Aβ42 downregulated MKP1 expression in PC12 cells
To assess the effect of Aβ42on the viability of PC12 cells, the cells were treated with different concentrations of Aβ42for 24 hours, and cell viability was assessed. Aβ42treatment resulted in the loss of cell viability in a dose-dependent manner (Figure 1A). To evaluate the effect of Aβ on MKP1 mRNA and protein expression in PC12 cells, 10 μM Aβ42was added to the cell culture medium, and MKP1 mRNA and protein expression was assessed by qRT-PCR and western blot assay at different time points. qRT-PCR showed that MKP1 mRNA expression was significantly downregulated 6 hours after Aβ42addition (P < 0.05), with the lowest expression at 12 hours(P < 0.01; Figure 1B). Western blot assay showed that MKP1 protein expression was significantly downregulated at 12 hours after Aβ42exposure (P < 0.01), with the lowest expression at 18 hours (P < 0.01) (Figure 1C). These results demonstrate that Aβ42downregulates MKP1 expression in PC12 cells in a time and concentration-dependent manner.
MKP1 suppressed Aβ42-induced oxidative stress
To examine the role of MKP1 in Aβ42-induced oxidative stress, we measured intracellular ROS levels in PC12 cells with the DCFH-DA assay. Aβ42treatment resulted in an increase in ROS generation compared with untreated control cells. In comparison, Aβ42treatment of PC12 cells with stable MKP1 knockdown with shRNA resulted in substantially higher ROS production compared with control cells. Conversely, cells with stable MKP1 overexpression showed reduced ROS generation compared with control cells (Figure 2A).
Mitochondrial membrane potential (ΔΨm) is a cellular indicator of oxidative stress, and ROS reduce ΔΨm (Vayssier-Taussat et al., 2002). We found that Aβ42exposure led to a reduction in ΔΨm in PC12 cells, which was significantly enhanced by MKP1 knockdown (P < 0.05), and significantly suppressed by MKP1 overexpression (P < 0.05; Figure 2B).SOD activity and MDA levels in PC12 cells were also measured. Aβ42treatment reduced SOD activity, and this effect of the peptide was significantly enhanced by MKP1 knockdown(P < 0.01) and significantly suppressed by MKP1 overexpression (P < 0.05; Figure 2C). In addition, Aβ42significantly increased MDA levels (P < 0.05). However, MKP1 knockdown had no impact on this Aβ42-mediated increase in MDA levels(P > 0.05), whereas MKP1 overexpression significantly diminished the Aβ42-mediated increase in MDA levels (P < 0.01;Figure 2D). These results demonstrate that MKP1 inhibits Aβ42-induced oxidative stress in PC12 cells.
MKP1 prevented Aβ42-induced neuroinflammation
To assess the effect of MKP1 on Aβ-induced neurotoxicity,PC12 cells were treated without (Control) or with 10 μM Aβ42(Aβ42) for 24 hours. Furthermore, PC12 cells stably expressing MKP1 shRNA (MKP1 KD + Aβ42) or MKP1 (MKP1 + Aβ)were treated with 10 μM Aβ42for 24 hours. phospho-JNK(p-JNK) levels were then measured by western blot assay, and mRNA levels of the inflammatory cytokines TNF-α and IL-1β were assessed by qRT-PCR. As shown in Figure 3A, Aβ42in-creased the p-JNK signal, and this effect was significantly enhanced by MKP1 knockdown (P < 0.05) and significantly diminished by MKP1 overexpression (P < 0.05). Aβ42increased mRNA levels of TNF-α and IL-1β, and MKP1 knockdown bolstered this effect of the peptide (P < 0.05). In contrast,MKP1 overexpression significantly diminished the Aβ42-mediated increase in expression of these inflammatory cytokines (P< 0.05; Figure 3B).
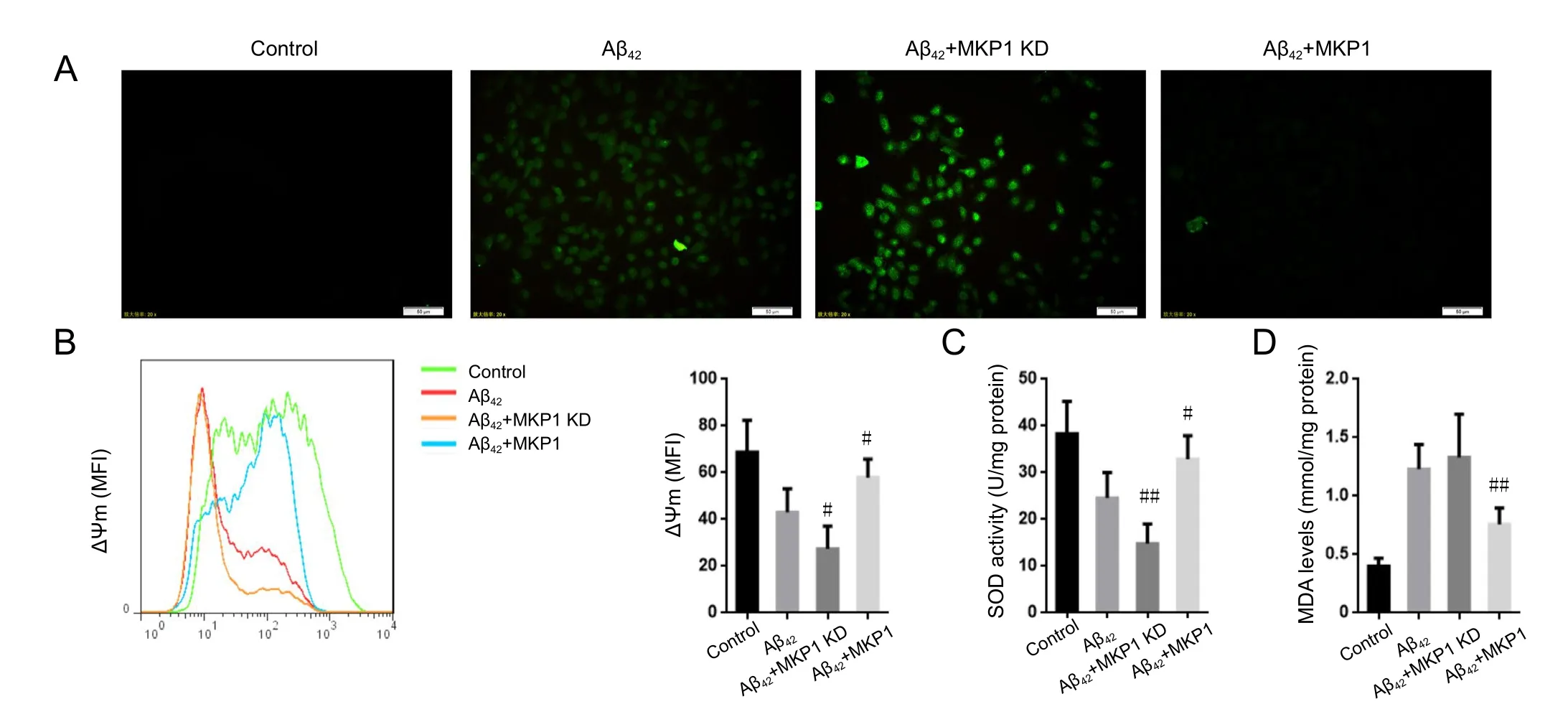
Figure 2 Eあect of MKP1 on Aβ42-induced oxidative stress in PC12 cells.
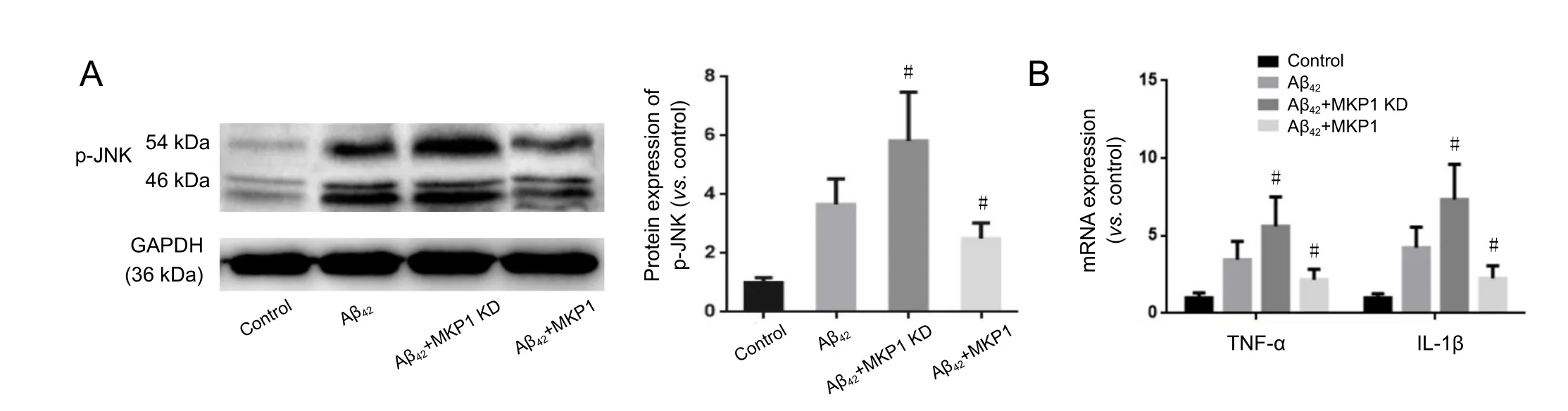
Figure 3 Eあect of MKP1 on Aβ42-induced neuroinflammation in PC12 cell culture.
MKP1 alleviated Aβ42-induced neuronal apoptosis
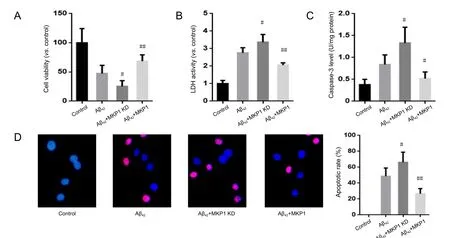
Figure 4 Eあect of MKP1 on Aβ42-induced neurotoxicity towards PC12 cells.
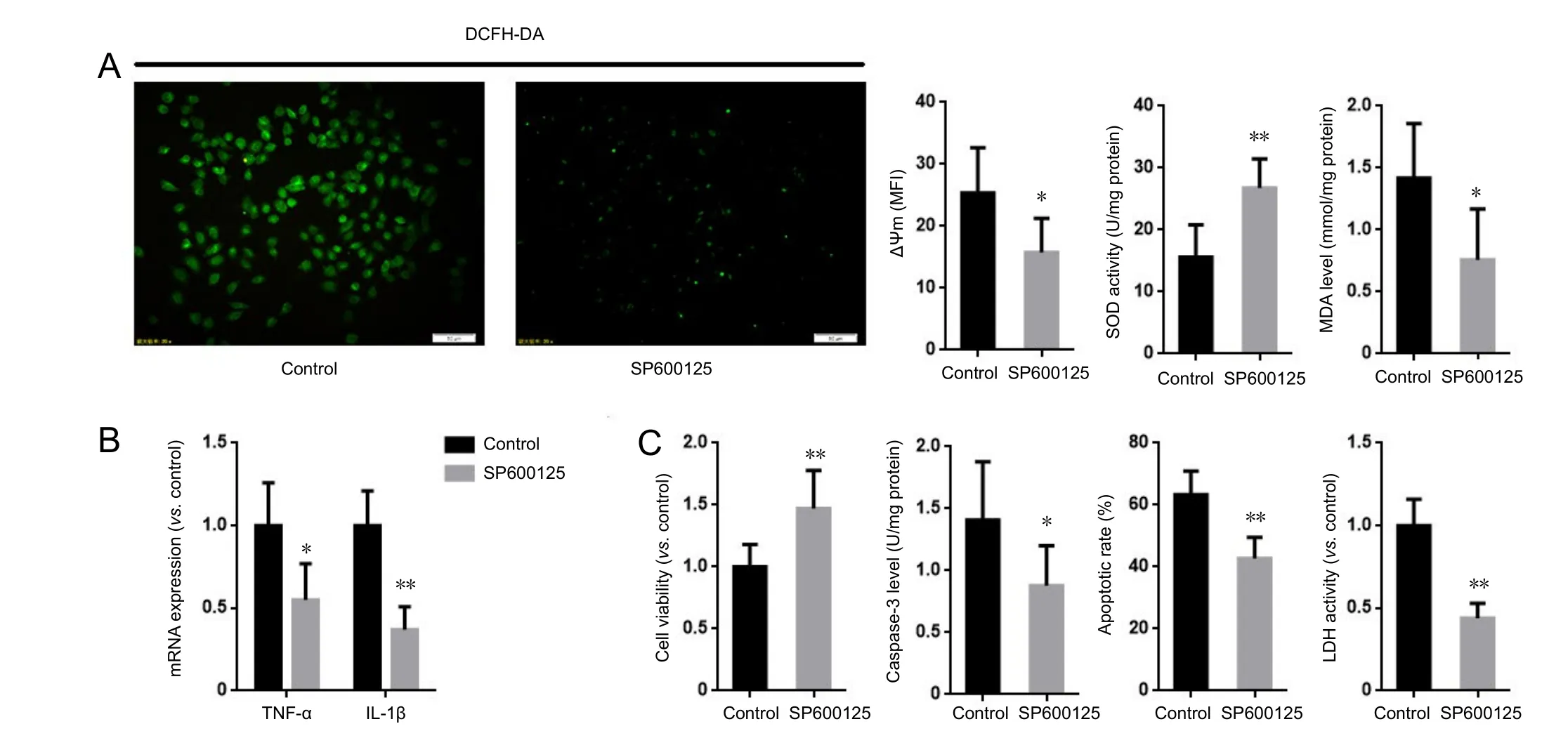
Figure 5 Inhibition of JNK suppressed the aggravating eあect of MKP1 knockdown on Aβ-induced neurotoxicity in PC12 cells.
To evaluate the effect of MKP1 on Aβ42-induced cellular injury, normal PC12 cells and PC12 cells stably expressing MKP1 shRNA or MKP1 were treated with 10 μM Aβ42for 24 hours,and cell viability was assessed with the cell counting kit-8 assay and by measuring the activity of LDH released into the medium. Aβ42reduced the viability of PC12 cells, and this effect of the peptide was significantly enhanced by MKP1 knockdown(P < 0.05) and significantly suppressed by MKP1 overexpression (P < 0.01; Figure 4A). Moreover, Aβ42increased LDH release. MKP1 knockdown enhanced Aβ42-induced LDH release(P < 0.05), whereas MKP1 overexpression diminished Aβ42-induced LDH release (P < 0.01; Figure 4B). In addition, Aβ42increased caspase-3 levels, which was significantly enhanced by MKP1 knockdown (P < 0.05) and significantly diminished by MKP1 overexpression (P < 0.05; Figure 4C). Furthermore,TUNEL staining demonstrated that Aβ42treatment led to the apoptosis of PC12 cells. The number of TUNEL-positive cells was significantly increased by MKP1 knockdown (P < 0.05)and significantly decreased by MKP1 overexpression (P < 0.01;Figure 4D).
Inhibition of JNK abolished the effect of MKP1 knockdown on Aβ42-induced neurotoxicity
The preceding results show that MKP1 attenuates Aβ42-induced oxidative stress, inflammation and cellular injury. To explore the role of the JNK pathway in the neuroprotective action of MKP1, PC12 cells stably expressing MKP1 shRNA were treated with or without the JNK-specific inhibitor SP6001250 (2 μM; Sigma-Aldrich) in the presence of 10 μM Aβ42for 24 hours, and cell viability, oxidative stress and apoptosis were assessed. SP600125 abrogated the MKP1 knockdown-induced increase in ROS generation and mitochondrial membrane potential. It also increased SOD activity and lowered MDA levels in PC12 cells (Figure 5A). In addition, SP600125 inhibited the MKP1 knockdown-induced upregulation of TNF-α and IL-1β mRNA expression (Figure 5B). Furthermore, SP600125 diminished the MKP1 knockdown-induced loss of cell viability, and alleviated the MKP1 knockdown-mediated increase in LDH release and apoptosis(Figure 5C).
Discussion
In the early phase of AD, Aβ accumulates in brain tissue and induces activation of MAPK signaling pathways, the generation of ROS and inflammatory reactions, ultimately leading to neuronal apoptosis (Origlia et al., 2009; Ghavami et al., 2014;Jazvinšćak Jembrek et al., 2015; González-Reyes et al., 2016;Jarosz-Griffiths et al., 2016). These phenomena have been verified in AD animal models and in vitro experiments; however,the mechanisms by which Aβ activates the MAPK signaling pathway remained unknown.
In this study, Aβ42neurotoxicity towards PC12 cells was concentration-dependent, and it decreased both MKP1 mRNA and protein expression. A previous study showed that MKP1 expression is downregulated in the brain tissue of patients with Huntington’s disease (Taylor et al., 2013), but it was unknown whether MKP1 expression is affected in AD.Our results revealed that MKP1 expression is downregulated following Aβ exposure, suggesting that MKP1 may be involved in the pathogenesis of AD.
There is substantial evidence that oxidative stress induced by ROS overproduction combined with the low antioxidative capacity of cells plays an important role in AD (Aliev et al., 2014;Luque-Contreras et al., 2014; Ganguly et al., 2017; Rojas-Gutierrez et al., 2017). Excessive oxidative stress leads to lipid peroxidation of the cell and organellar membranes, thereby affecting the function of nerve cells (Shichiri, 2014; Di Domenico et al., 2017). Moreover, oxidative stress results in nitration of proteins and damage to nucleic acids (Stepien et al., 2017;Wu and Tang, 2018). These combined negative effects eventually lead to the functional impairment or loss of neurons.Aβ catalyzes the generation of ROS via the α-helical structure,with evidence demonstrating that the oxidative stress in the brains of AD patients and animal models is mainly produced by Aβ (Prasad and Bondy, 2014). In this study, Aβ induced the generation of ROS in PC12 cells. Intriguingly, ROS generation in PC12 cells was substantially increased after MKP1 knockdown, while it was abolished by MKP1 overexpression.Moreover, ROS reduce ΔΨm, a biological indicator of oxidative stress. We found that MKP1 knockdown enhanced, while MKP1 overexpression inhibited the Aβ42-induced decrease in ΔΨm. In addition, MKP1 knockdown enhanced, while MKP1 overexpression inhibited the Aβ42-induced decrease in SOD activity in PC12 cells. Furthermore, MKP1 overexpression prevented the increase in MDA levels induced by Aβ42. These results indicate that MKP1 plays an important role in Aβ-induced oxidative stress in neurons.
Chronic inflammation in the brain is another important pathological feature of AD (Bagyinszky et al., 2017; Fraga et al., 2017; Shamim and Laskowski, 2017). Epidemiological studies have shown that nonsteroidal anti-inflammatory drugs significantly reduce the incidence of AD, and nonsteroidal anti-inflammatory drugs and corticosteroids reduce Aβ deposition and suppress the activation of glial cells, as well as the release of inflammatory factors and free radicals in the brains of AD animal models (Doost Mohammadpour et al., 2015;Heneka et al., 2015). JNK plays an important role in inflammation (Cano and Mahadevan, 1995) and is involved in the regulation of transcription of inflammatory genes via phosphorylation of the downstream target c-Jun (Coffey, 2014). In this study, we demonstrated that Aβ promotes JNK activation.Furthermore, MKP1 modulated the Aβ-induced activation of the JNK pathway, accompanied with changes in the expression of the inflammatory cytokines TNF-α and IL-1β. JNK is also involved in apoptosis (Chen, 2012), and the inhibition of the JNK pathway alleviates Aβ-induced neuronal apoptosis(Bozyczko-Coyne et al., 2001). In this study, the JNK specific inhibitor SP600125 prevented the MKP1 knockdown-induced increase in ROS generation. It also reduced oxidative stress and the generation of inflammatory factors, and it prevented apoptosis caused by Aβ. These findings suggest that JNK modulates the action of MKP1 in Aβ-induced toxicity.
There are some limitations to this study. The mechanisms by which Aβ downregulates MKP1 need to be further clarified. Furthermore, an in vivo study should be conducted to evaluate the neuroprotective effects of MKP1 in AD.
In summary, Aβ downregulates MKP1 expression. MKP1,in turn, alleviates Aβ-induced oxidative stress, inflammation and cellular injury by suppressing the JNK signaling pathway.Our findings provide insight into the mechanisms by which the MAPK signaling pathway is activated by Aβ. The MKP1/JNK signaling axis may be a promising therapeutic target for the development of novel drugs for AD.
Author contributions: YG, LJM and XPL conceived and designed the study. JJ, XFZ and DC performed the experiments. YG and LJM wrote the paper. XPL and XXB reviewed and edited the paper. All authors read and approved the final version of the paper.
Conflicts of interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- Validity and reliability of the Ocular Motor Nerve Palsy Scale
- High-frequency (50 Hz) electroacupuncture ameliorates cognitive impairment in rats with amyloid beta 1–42-induced Alzheimer’s disease
- Kaempferol attenuates cognitive deficit via regulating oxidative stress and neuroinflammation in an ovariectomized rat model of sporadic dementia
- Combined VEGF/PDGF improves olfactory regeneration after unilateral bulbectomy in mice
- Comparison of morphological and functional outcomes of mouse sciatic nerve repair with three biodegradable polymer conduits containing poly(lactic acid)
- Transcriptome analysis of adherens junction pathwayrelated genes after peripheral nerve injury