
Determination of 11 synthetic musks in imported seafood by solid phase extraction and gas chromatography-mass spectrometry
2018-08-02 10:33QULiZENGJingZHAOChaominSONGWeimin
色谱 2018年8期
QU Li, ZENG Jing, ZHAO Chaomin, SONG Weimin
(1. Shanghai Entry-Exit Inspection and Quarantine Bureau, Shanghai 200135, China; 2. School of Public Health, Fudan University, Shanghai 200032, China)
Abstract: A method has been developed for the simultaneous determination of 11 synthetic musks (cashmeran, celestolide, phantolide, musk ambrette, traseolide, galaxolide, musk xylene, tonalide, musk moskene, musk tibetene, and musk ketone) in imported seafood. The method combines solid phase extraction and gas chromatography-mass spectrometry. Samples were extracted by n-hexane and purified using a Florisil column. Internal standards were used to correct for matrix effects. The calibration curves showed good linearity with correlation coefficients greater than 0.99. The limits of detection (S/N>3) ranged from 0.35 to 2.08 μg/kg, and the limits of quantification (S/N>10) were between 1.18 and 5.00 μg/kg. The average recoveries measured at three spiked levels (10, 20, and 30 μg/kg) were in the range 83.1%-117% , with relative standard deviations ranging from 5.1% to 8.5% . Further, the concentrations of 11 synthetic musks in 30 imported seafood in Shanghai were investigated. Galaxolide was detected in 93.3% of samples analysed, in concentration as high as 3.82 μg/kg. Musk ambrette and musk moskene were found in concentrations as high as 15.4 μg/kg and 10.5 μg/kg, respectively. The established method demonstrates high sensitivity and selectivity for the determination and confirmation of 11 synthetic musks in imported seafood.
Key words: solid phase extraction (SPE); gas chromatography-mass spectrometry (GC-MS); synthetic musks; imported seafood
Synthetic musks are important ingredients widely utilized in consumer products such as perfumes, cosmetics, soaps, shampoos, detergents, fabric conditioners, cleaning products, and air fresheners, due to their typical musky scents and fixative properties. According to their chemical structures, synthetic musks are classified into nitroaromatic musks (musk ambrette, musk moskene, musk tibetene, musk ketone, and musk xylene), polycyclic musks (celestolide, galaxolide, tonalide, phantolide, cashmeran, and traseolide), and macrocyclic musks [1]. Synthetic musks have been identified as posing potential risks to the environment based on information regarding persistence, accumulation in organisms, and the potential to cause harm to organisms. Many countries have assessed the potential risk of synthetic musks. For example, the Canadian Environmental Protection Act clearly stipulates a reduction in the use of synthetic musks [2]. China and other countries have issued maximum residue limits (MRLs) for synthetic musks based on the European Union Cosmetic Regulation No.1223/2009 [3]. In most of the current laws and regulations, three synthetic musks are specifically prohibited (musk ambrette, musk tibetene, and musk moskene) because of their high toxicity and sensitisation, while the concentrations of four other synthetic musks are limited in cosmetics (musk ketone, musk xylene, phantolide and tonalide).
Seafood is not only one of the most-consumed foods worldwide, but is also one of the strongestbioaccumulators. Since synthetic musks have lipophilic characteristics and slow biodegradation, they can be biomagnified through the food chain[4]. Both larger and older fish have a greater potential of being contaminated with synthetic musks [5]. With the increase in consumption of seafood, this may lead to increased human exposure, especially for coastal people. In the higher-tier assessment, attention has also been paid to the potential risks posed to human health and the environment [6-8].
Many methods have been used to detect synthetic musks in different matrices, including environmental samples and personal care products [9-13]. Musks have been detected in water [14,15], wastewater [16,17], sewage [18,19], house dust [20-22], breast milk [23], and even in snow [24]. Previous studies have even confirmed the presence of synthetic musks in fish [25-29]. However, these previous studies focused on fewer target compounds or fewer kinds of seafood matrices. There is thus a lack of research on the determination of synthetic musks in seafood. Synthetic musk concentrations in imported seafood products in Shanghai, China, has never been reported before.
The aim of this work was to develop an accurate and sensitive method for the detection of 11 synthetic musks (cashmeran, celestolide, phantolide, musk ambrette, traseolide, galaxolide, musk xylene, tonalide, musk moskene, musk tibeten, and musk ketone) in seafood. The method was based on solid phase extraction (SPE), which can effectively remove impurities, leading to improved purification and reduced matrix effects. Tandem MS was selected to enhance selectivity and decrease the limits of detection (LOD). The residual content was determined and confirmed by GC-MS and quantified by an internal standard method.
1 Experimental
1.1 Apparatus and chemicals
All reagents were analytical grade unless otherwise mentioned. A total of 11 synthetic musks in cyclohexane, and musk xylene-D15in acetone, were purchased from Dr. Ehrenstorfer (Augsburg, Germany). Table 1 displays the names, abbreviations, CAS numbers, molecular formulas, purities, retention times, and MS conditions for all synthetic musks. To prevent volatilisation, the standard solutions were stored in capillary bottles (Certan, Germany). Stock solutions were prepared in acetone and stored at 4 ℃ in the dark. Musk-free plastic tubes were used in our study in order to avoid the complexities of handling glassware.

Table 1 Chemical names, CAS numbers, molecular formulas, purities, log Kow, retention times, quantification and identification ions of GC-MS
* The data in brackets are relative abundance.
HPLC-grade acetone (99.9% ), dichloromethane (99.9% ), andn-hexane (99.9% ) were obtained from Merck (Darmstadt, Germany) and used without further purification. All water used was purified with an Integral-10 Milli Q water purification system (Bedford, MA, USA).
The instruments used included a high-speed tissue crusher (Grindomix GM 200, Retsch, Germany), analytical balance (LP620P, Sartorius, Germany), vortex mixer (Scientific Industries, USA), centrifuge (Allegra X-30R, Beckman, USA), nitrogen evaporator (Turbo Vap, Caliper Lifesciences, Germany), a SPE set (12-position, Supelco, USA). The SPE column used in the experiment was LC-Florisil SPE column (1 g, 6 mL, Supelco, USA).
1.2 Sample preparation
All seafood samples were purchased from supermarkets in Shanghai, China. Shellfish such as lobster, shrimp, crab, oysters, and mussels were first skinned and de-shelled before proceeding. The edible parts of representative seafood samples were smashed thoroughly by a tissue crusher and put in clean containers to avoid contamination or any other factors that may have affected the sample composition. The test samples were stored at -18 ℃ before analysis.
1.3 Sample extraction and purification
Samples were removed from the freezer and allowed to return to room temperature. A total of 2.0 g seafood tissue was accurately weighed and placed in a 50 mL plastic centrifuge tube, to which 20 ng of internal standard (Musk xylene-D15) was added. Subsequently, 5 mLn-hexane was added, and the centrifuge tube was subjected to ultrasonic bath for 10 min and then centrifuged for 5 min at 4 000 r/min. Finally, the supernatant was transferred into a new plastic centrifuge tube. The extraction procedure was repeated one time, and the supernatants were combined. The supernatant was evaporated to a final volume of approximately 2 mL under a gentle stream of nitrogen at room temperature.
The Florisil column was sequentially rinsed with 5 mL of dichloromethane and 5 mL ofn-hexane prior to use. The sample solution was transferred into the column, and the effluent was discarded. The column was then eluted with 6 mL of dichloromethane at a flow rate of 1 drop/s. All eluate was collected in a 15 mL clean plastic tube and evaporated to dryness under a gentle stream of nitrogen. Finally, the residue was dissolved inn-hexane and diluted to 1.0 mL before being filtered through a 0.22 μm polytetrafluoroethylene (PTFE) filter for GC-MS determination and confirmation.
1.4 GC-MS analysis
GC-MS separation was performed with an Agilent 7890B gas chromatograph coupled with an Agilent 5977A mass spectrometer (Agilent Technologies, USA). The system was operated using the MassHunter software and NIST library search. An HP-5MS ([5%-phenyl]-methylpolysiloxane) fused silica capillary column (length 30 m, internal diameter 0.25 mm, film thickness 0.25 μm) was obtained from Agilent Technologies (USA). Helium (purity≥ 99.999% ) was used as a carrier gas at a constant flow rate of 1.0 mL/min. The temperature for the GC oven was initially set to 60 ℃ (where it was held for 1 min), increased to 190 ℃ at 40 ℃/min (where it was held for 6 min), and finally increased to 220 ℃ at 20 ℃/min (where it was held for 2 min). The experiment was conducted in splitless injection mode, and run time was 13.75 min. A solvent delay of 5.0 min was selected. The MS detector was operated in selected ion monitoring (SIM) mode, and injector temperature was maintained at 230 ℃. The transfer line temperature was 250 ℃. Retention times as well as characteristic quantification and identification ions for each target compound are summarised in Table 1.
Positive samples were confirmed by GC-MS/MS. Analysis was performed on an Agilent 7000A triple quadrupole mass spectrometer and an Agilent 7890A gas chromatograph equipped with a 30 m×0.25 mm, 0.25 μm HP-5MS ([5%-phenyl]-methylpolysiloxane) fused silica capillary column (Agilent Technologies, USA). The system was operated with the MassHunter software and NIST library search. Helium (purity≥ 99.999% ) was used as a carrier gas at a constant flow rate of 1.0 mL/min. A total of 1.0 μL of each sample was injected in splitless mode at 280 ℃. The temperature program for the GC oven began at 60 ℃ where it was held for 1 min, then increased to 160 ℃ at 20 ℃/min, and finally increased to 166 ℃ at 2.0 ℃/min.
The MS/MS analysis was carried out in electron ionization (EI) mode. Multiple reaction monitoring (MRM) mode was utilized for quantitative analysis of target compounds. The temperature of the injector and transfer line were kept at 230 ℃ and 250 ℃, respectively. Solvent delay was 5.0 min. The selected monitoring ions for each compound were optimized and are listed in Table 2 along with their retention times.
2 Results and discussion
2.1 Optimization of extraction conditions and extraction time
Extraction solvent and extraction time are the primary factors affecting extraction efficiency from the seafood samples. Therefore, the optimal solvents were first selected through a preliminary experiment based on the principle of dissolution of similar polarity compounds. The logKowvalues of the synthetic musks (Table 1) indicated that these compounds are highly lipophilic, so an organic solvent with weak polarity was selected for extraction. The 11 synthetic musk standards were purchased as solutions in cyclohexane. Based on the principle of similar phase dissolution and the polarity of the compound, it is easy to choose a suitable solvent for extraction. The polarity of acetone isP=5.1 which is highly polar compared ton-hexane (P=0.1), so acetone would not be suitable for the extraction of polar synthetic musks. Thereforen-hexane was chosen as the extraction solvent [29].
In order to obtain the best recovery, ultrasonic extraction was compared with vortex extraction with different durations (5, 10, and 15 min) using the spiked seafood samples. Fig. 1 demonstrates that the recoveries of the 11 synthetic musks reached their maximum values (over 80% ) after ultrasonic extraction for 10 min. Therefore, ultrasonic extraction for 10 min was used to extract the target analytes from samples.
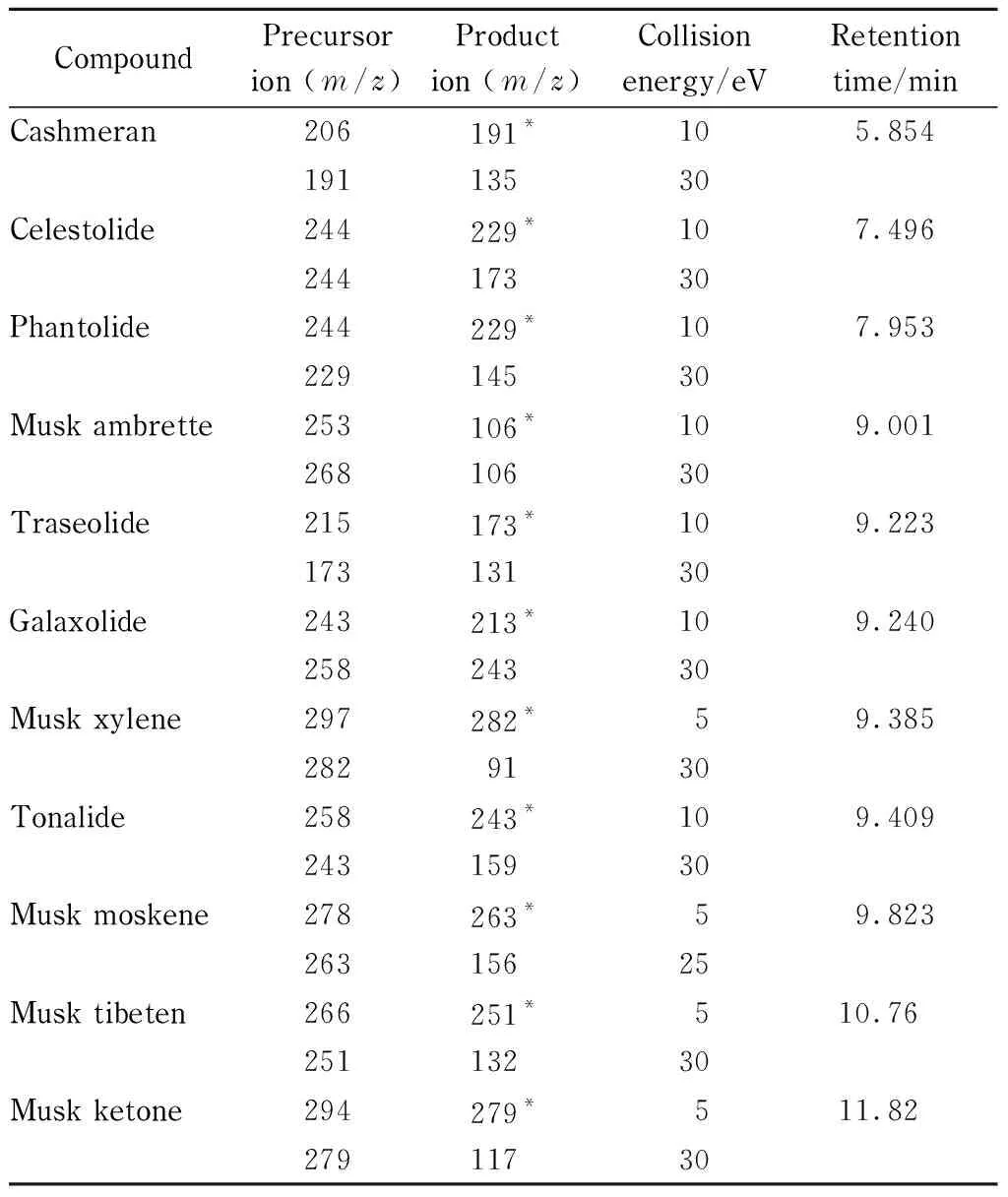
Table 2 Monitoring ions and retention times of 11 synthetic musks by GC-MS/MS
* Quantitative ion.
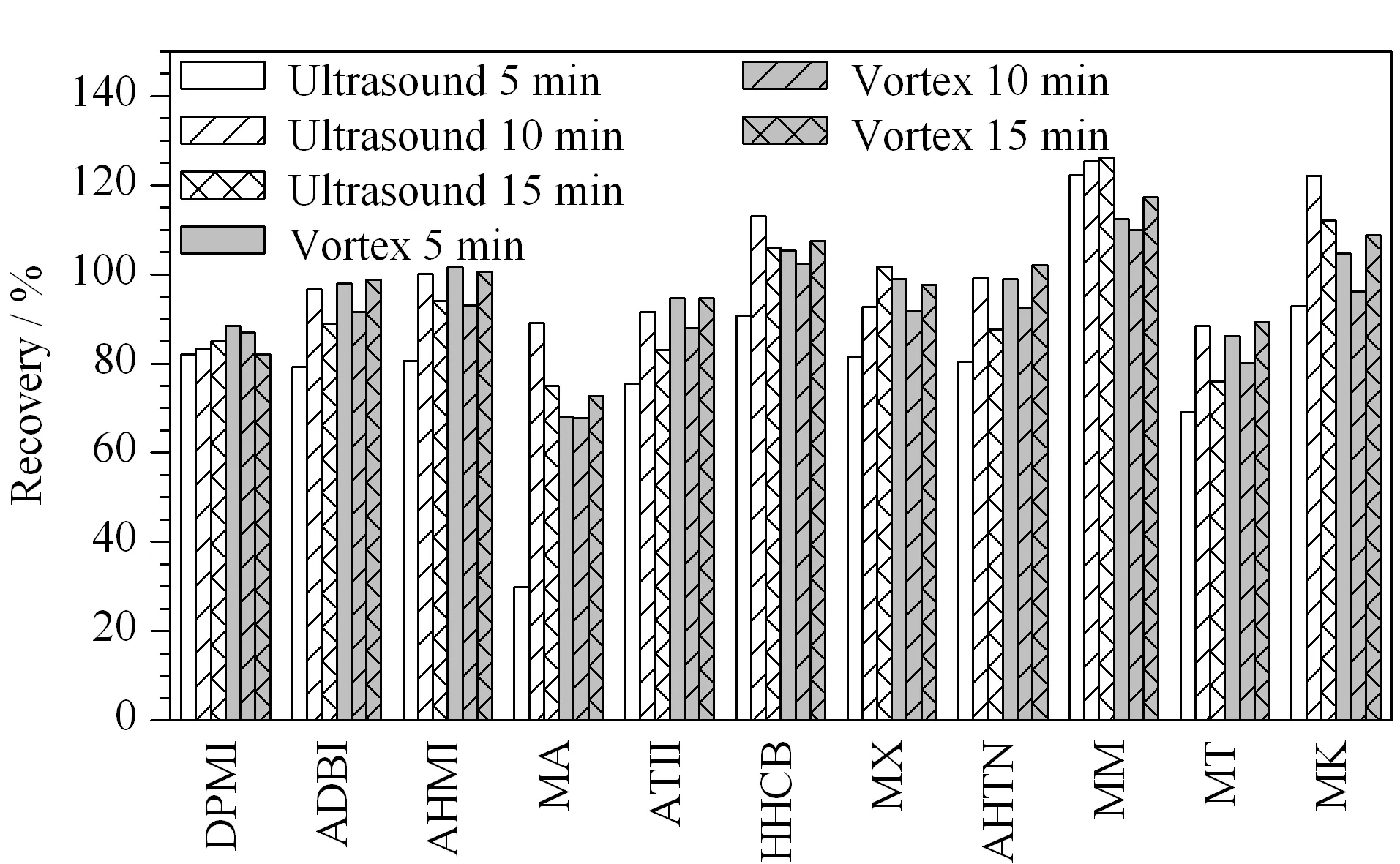
Fig. 1 Comparison of recoveries from samples spiked with 10 ng/g target analytes, based on different extraction methods and times
2.2 Selection of the SPE column
Since synthetic musks are weakly polar compounds, we used normal-phase extraction to remove polar impurities. In this study, four types of SPE columns, including LC-C18SPE (500 mg, 6 mL, Agela, USA), HLB SPE (500 mg, 6 mL, Waters, USA), LC-Florisil SPE (1 g, 6 mL), and LC-Si SPE (500 mg, 6 mL, Supelco, USA), were compared through evaluation of the recoveries of 11 synthetic musks from spiked seafood samples. Fig. 2 demonstrates that the recoveries of 11 synthetic musks were nearly 80% when using an LC-Florisil SPE column. The recoveries when using the LC-Florisil SPE column were higher than those obtained with the LC-Si SPE. Therefore, the LC-Florisil SPE column was selected as the best among the four SPE columns tested.

Fig. 2 Comparison of recoveries from seafood samples spiked with 10 ng/g target analytes, using different SPE columns
Florisil is a highly selective adsorbent with a strong ability to remove impurities, and on which synthetic musk compounds have high adsorb abilities. It can achieve a satisfactory recovery with lower background noise and higher sensitivity.
2.3 Optimization of eluent
Different organic reagents (n-hexane, cyclohexane, dichloromethane, and ethyl acetate) were screened. Dichloromethane demonstrated the best elution efficiency among all eluents tested, completely eluting all 11 synthetic musks, and it was generally applicable to different matrices.
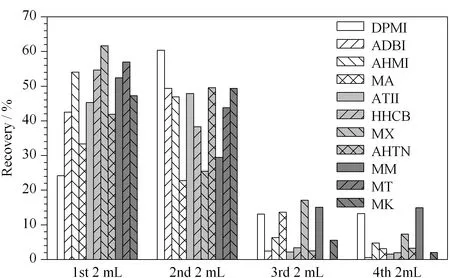
Fig. 3 Relationship between recovery and elution volume for the 11 synthetic musks
When the target compounds were eluted from the LC-Florisil SPE column, the 1st, 2nd, 3rd, and 4th 2 mL dichloromethane fractions were collected in different tubes and then evaporated to dryness under a gentle stream of nitrogen in order to obtain good recoveries and use of the least amount of solvent in the experiment. Fig. 3 displays the relationship between the recovery and elution volume for the 11 synthetic musks. The results suggested that the 11 synthetic musks could be completely eluted in a volume of 8 mL, with recoveries ranging from 81.9% to 101% except for musk ambrette (56.1% ). The recovery of musk ambrette was increased to 95.3% using an elution volume of 8 mL.
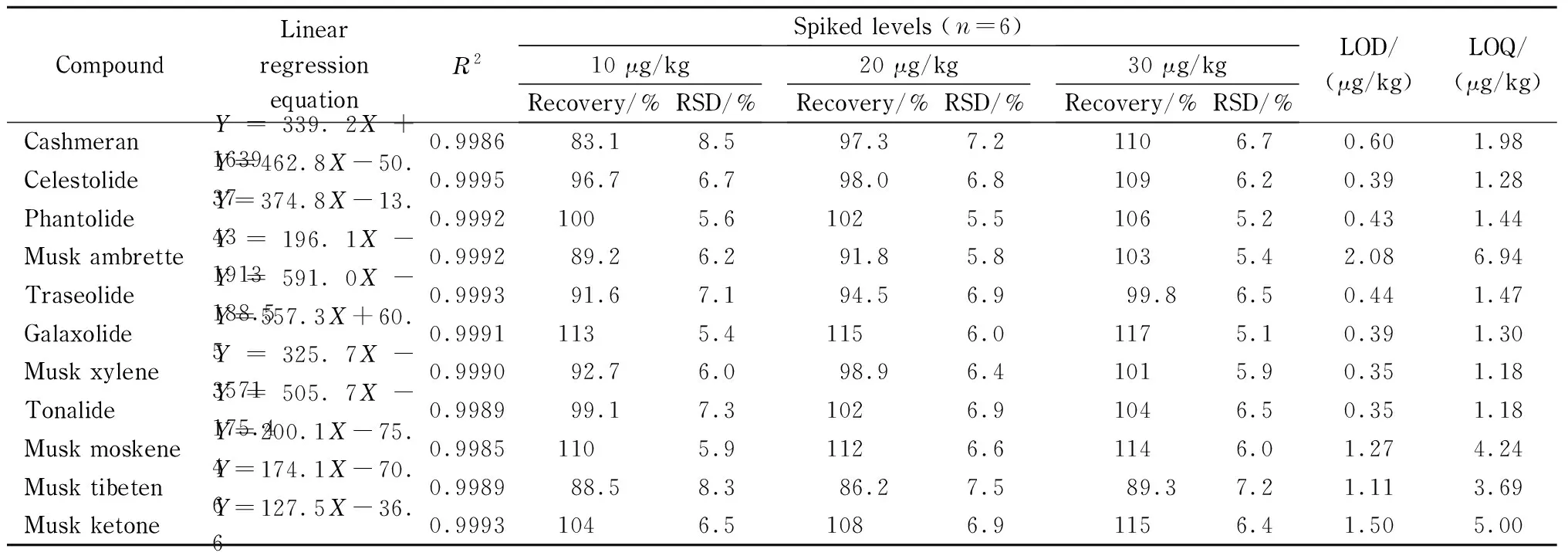
Table 3 Linear relationships, recoveries, LODs and LOQs of the 11 synthetic musks spiked in imported seafood
Y: peak area of target compound;X: mass concentration of target compound, mg/L.
2.4 Method validation
To assess accuracy and precision, three concentrations of mixed standard solutions were spiked into a seafood sample without targets and prepared for analysis according to the aforementioned procedure. Six replicates were performed for each concentration. The average recoveries were above 83.1% after correcting with internal standards (Table 3), which was satisfactory. The relative standard derivations (RSD) were lower than 9.0% . The calibration solutions ranging from 1 μg/L to 100 μg/L (including 50 μg/L of internal standard) resulted in acceptable linearity for all compounds (correlation coefficients (R2)> 0.99). The LODs (S/N>3) were 0.35-2.08 μg/kg, and the limits of quantification (LOQs,S/N>10) were 1.18-5.00 μg/kg, which are better than those of a previously reported method [23]. Fig. 4 shows a GC-MS chromatogram of a standard solution (50 μg/L) inn-hexane. There was a small degree of overlap between the peaks of AHTN and MX, but they could be distinguished by extracting the specific ions, AHTN (m/z243) and MX (m/z282). MRM chromatograms for all 11 synthetic musks are shown in Fig. 5.
2.5 Analysis of seafood samples
The newly developed method was applied in the analysis of 30 imported seafood samples purchased from local markets in Shanghai, China.Synthetic musks were detected in 100% of the analysed samples. The musks most widely detected were MA, HHCB, and MM, in concentrations up to 15.4 μg/kg. Table 4 confirms that both of the restricted musks, MA and MM, were detected in seafood samples in concentration ranges of 7.93-15.4 ng/g and 5.43-10.5 ng/g, respectively. MA was found in 86.7% of analysed samples, although in 53.3% the concentration was lower than the LOQ. Ninety percent of analysed seafood samples contained detectable levels of MM; 30% had concentrations below the LOQ. Although in low levels, HHCB was also found in most samples in a concentration range of 1.30-3.82 ng/g. HHCB was the most commonly detected substance (in 93.3% of samples), probably because HHCB was one of the most widely used synthetic musks worldwide, resulting in accumulation in seafood through biomagnification. Concentrations of HHCB in seafood samples from the Shanghai Port were lower than the values reported in samples from ports in Europe and Spain [22,23]. However, the two studies referenced were limited in their analyses of only single species in given samples. In this study, more types of seafood were investigated, and more synthetic musks were monitored.

Fig. 4 GC-MS total ion current chromatogram of a 50 μg/L standard solution of 11 synthetic musks in n-hexane

N. D.: not detected
3 Conclusions
In this study we developed and optimized an accurate and reliable method for the determination of 11 synthetic musks in seafood samples. The method allows for rapid and simultaneous determination of nitro musks and polycyclic musks in imported seafood, with high precision, accuracy, and low LODs. The method includes extraction using a Florisil SPE column and subsequent GC-MS analysis. The application of tandem mass spectrometry not only improved the sensitivities toward nitro musks and polycyclic musks, but also eliminated matrix interferences. Musk xylene-D15could be applied to eliminate the matrix effects. Moreover, a comprehensive assessment of the levels of synthetic musks in seafood imported into the Shanghai Port was performed; two globally prohibited synthetic musks (MA and MM) as well as HHCB were detected in a large proportion of samples at low concentrations. These compounds could be accumulated to even higher levels in humans, which may negatively affect human health.
AcknowledgementThe authors thank Dr. DENG Xiaojun for technical assistance and material support.
