
离子液体分散液液微萃取-超高效液相色谱-串联质谱法测定食品接触材料中全氟辛酸和全氟辛烷磺酸的迁移量
2018-08-02 10:32闫萌萌孟宪双郭项雨王鹏龙雷海民
色谱 2018年8期
闫萌萌, 陈 萌, 孟宪双, 郭项雨,白 桦*, 马 强*, 王鹏龙, 雷海民
(1. 中国检验检疫科学研究院, 北京 100176; 2. 北京中医药大学中药学院, 北京 100029)
食品包装是食品的重要组成部分,具有保护食品不受外来生物、化学和物理因素的破坏,维持食品质量稳定的特点。食品接触材料是指在正常使用条件下,各种已经或预期可能与食品或食品添加剂接触或其成分可能转移到食品中的材料和制品。由于食品接触材料会与食品直接接触,因而对食品安全有着双重影响:一方面质量合格的食品接触材料可以保护食品不受外界的污染,保持食品的水分、成分、品质等特征不发生改变;另一方面食品接触材料的原材料在生产过程中可能受到污染,且在加工处理过程中需要加入各种功能性助剂,这些成分可能会向其接触的食品中迁移[1],这些化学物质迁移在食品中达到一定量时可能危害消费者的健康。
在各种受到关注的化学物质中,全氟化合物(PFCs)尤为受到关注。全氟化合物是化合物分子中与碳原子连接的氢原子全部被氟原子所取代的一类有机化合物,主要包括全氟羧酸类化合物(PFCAs)、全氟磺酸类化合物(PFSAs)等;其中,全氟辛酸(PFOA)和全氟辛烷磺酸(PFOS)是两种最为常见、用量最大的全氟化合物[2-4]。由于全氟化合物具有化学性质稳定、疏水疏油、耐高温、表面活性强等特性,被广泛应用于食品包装、纺织、皮革、电子产品、洗涤用品等工业与消费品领域[2,3,5,6]。全氟化合物具有较强的生物富集性、持久性,已有的毒理学研究表明,全氟化合物具有生殖毒性、神经毒性、免疫毒性等多种毒性,被认为是一类具有全身多脏器毒性的环境污染物,严重威胁人类健康[4,5,7-9]。
包装纸、包装袋及各类涂层等食品接触材料中的全氟化合物可能迁移至食品中或在烹饪过程中释放[10,11],进而对消费者的健康安全构成潜在危害。目前,关于食品接触材料中全氟化合物检测的文献报道主要关注残留量测定,采用的方法主要为气相色谱(GC)法[12]、气相色谱-质谱(GC-MS)法[13,14]、气相色谱-三重四极杆质谱(GC-MS/MS)法[15]、高效/超高效液相色谱-三重四极杆质谱(HPLC/UPLC-MS/MS)法[16-18]、高效/超高效液相色谱-四极杆-飞行时间质谱(HPLC/UPLC-Q-TOF-MS)法[19-21]等。以上研究均为针对食品接触材料本身,关于食品接触材料中全氟化合物迁移量测定的文献报道相对较少[22-25]。对食品接触材料中化学物质的迁移行为进行关注、研究、测试和控制,对保证食品安全、保护消费者健康具有非常重要的意义。目前消费者普遍关注的是食品接触材料在推荐的使用条件下与食品接触时,迁移到食品中的物质水平是否符合安全限量的要求,这一符合性评估一般通过迁移试验方法测定实际迁移量。以往迁移试验中大体积溶液处理方法主要有液液萃取法[26-28]、固相萃取法[29-31]等。液液萃取法设备简单,但操作繁琐、耗时较长,有机溶剂消耗量大,选择性较差,适合于高浓度目标物的分离;固相萃取法相比液液萃取法具有更高的准确率和更稳定的回收率,但操作繁琐,需要活化、上样、淋洗、洗脱,耗时长,价格成本高。
针对上述迁移溶液处理中的问题,本研究采用离子液体分散液液微萃取技术,对食品接触材料迁移溶液中的PFOA和PFOS进行富集,并对影响萃取效果的主要因素(萃取剂的种类与用量、涡旋时间、无机盐含量、离心转速与时间等)进行了优化,结合超高效液相色谱-串联质谱法测定迁移量,建立了一种离子液体分散液液微萃取-超高效液相色谱-串联质谱法测定食品接触材料中全氟辛酸和全氟辛烷磺酸迁移量的分析方法。
1 实验部分
1.1 仪器与试剂
ACQUITY超高效液相色谱仪、XEVO TQ三重四极杆质谱仪、MassLynx数据处理系统(美国Waters公司); Milli-Q超纯水器(美国Millipore公司); AB204-S型电子天平(瑞士Mettler Toledo公司); HH-1数显恒温水浴锅(常州万合仪器制造有限公司); PFOA标准品(CAS No. 335-67-1,纯度为98% )购自美国Sigma-Aldrich公司,PFOS标准品(CAS No. 1763-23-1,纯度99% )购自德国Dr. Ehrenstorfer公司;1-丁基-3-甲基咪唑六氟磷酸盐([C4MIM][PF6], CAS No. 174501-64-5,纯度99% )和1-己基-3-甲基咪唑六氟磷酸盐([C6MIM][PF6], CAS No. 304680-35-1,纯度99% )购自百灵威科技有限公司;甲醇和乙腈(均为色谱纯)购自美国Fisher公司;乙酸、乙醇、氯化钠和乙酸铵(均为分析纯)购自北京化工厂。
1.2 标准储备液及标准工作溶液的配制
准确称取全氟辛酸和全氟辛烷磺酸标准品各10.0 mg,用甲醇溶解并定容至10 mL,配制成1 g/L标准储备液,于4 ℃保存。使用时用甲醇逐级稀释成系列质量浓度的标准工作溶液,现用现配。
1.3 迁移试验
将食品接触材料试样剪碎至5 mm×5 mm以下,按照食品安全国家标准GB 5009.156-2016和GB 31604.1-2015的要求进行迁移试验。向水、10%(体积分数,下同)乙醇溶液和4%(体积分数,下同)乙酸溶液3种水性食品模拟物试液中分别加入0.5 g食品接触材料试样。在70 ℃恒温水浴条件下浸泡12 h后,移取5 mL迁移液于15 mL具塞离心管中,向离心管中依次加入220 μL离子液体[C6MIM][PF6]和5 mL 6%(质量分数,下同)氯化钠溶液,经涡旋提取3 min后,以5 000 r/min的速度离心3 min,移取下层液体22 μL,用甲醇稀释至1 mL,供超高效液相色谱-串联质谱测定。
1.4 分析条件
色谱柱:ACQUITY UPLC BEH Shield RP18(50 mm×2.1 mm, 1.7 μm);流速:0.4 mL/min;流动相A为水,流动相B为乙腈。梯度洗脱程序:0~4.0 min, 20%B~80%B; 4.0~5.0 min, 80%B; 5.0~5.1 min, 80%B~20%B; 5.1~6.0 min, 20%B;柱温:40 ℃;进样量:5 μL。
电喷雾离子源负离子扫描模式;毛细管电压:2.4 kV;射频透镜电压:0.5 V;离子源温度:120 ℃;脱溶剂气温度:500 ℃;脱溶剂气流量:1 000 L/h;碰撞气:氩气;碰撞气压:0.32 Pa;数据采集模式:多反应监测(MRM)。PFOA和PFOS的质谱分析参数见表1, MRM色谱图见图1。

表 1 全氟辛酸和全氟辛烷磺酸的质谱参数Table 1 MS parameters of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS)
* Quantitative ion.
2 结果与讨论
2.1 离子液体种类和用量的选择
分别考察了[C4MIM][PF6]和[C6MIM][PF6]两种咪唑类离子液体对3种食品模拟物中PFOA和PFOS萃取效果的影响(见图2a)。在水、10%乙醇溶液和4%乙酸溶液中,[C6MIM][PF6]对PFOA和PFOS的萃取回收率均接近100%(94.0% ~112.1% ),较稳定;在使用[C4MIM][PF6]进行萃取时,回收率为84.3% ~146.4% ,波动较大。而且在10%乙醇溶液中,PFOA在[C4MIM][PF6]萃取下的回收率(84.3% )较[C6MIM][PF6](101.2% )低,因此,综合考虑后选择[C6MIM][PF6]作为3种食品模拟物的萃取剂。
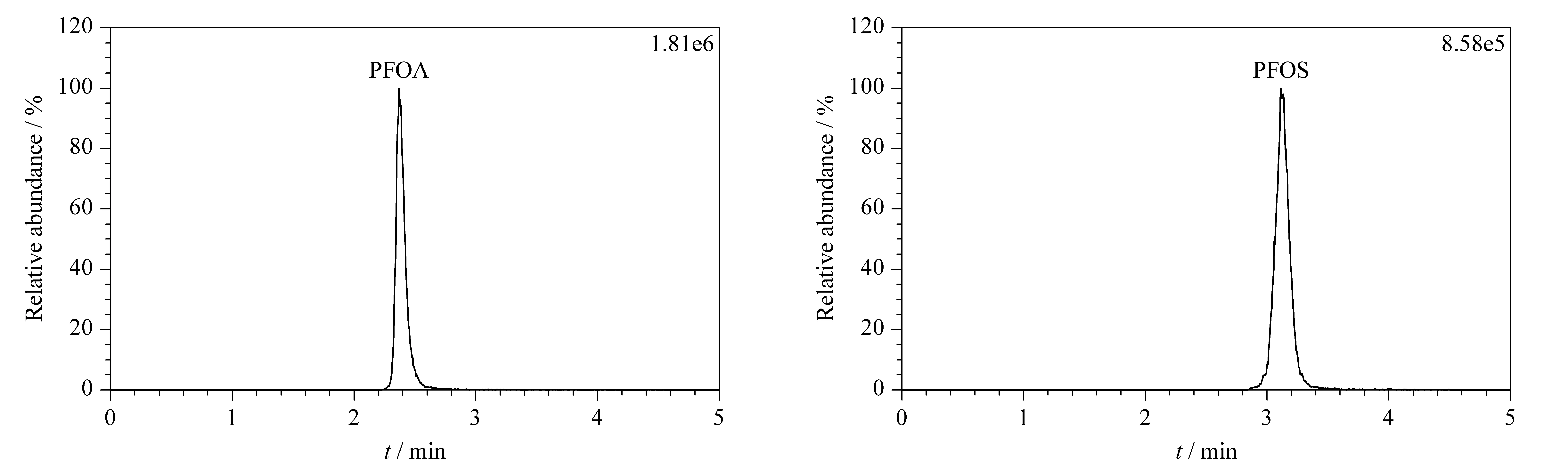
图 1 PFOA和PFOS标准溶液的多反应监测色谱图Fig. 1 MRM chromatograms of PFOA and PFOS standard solutions
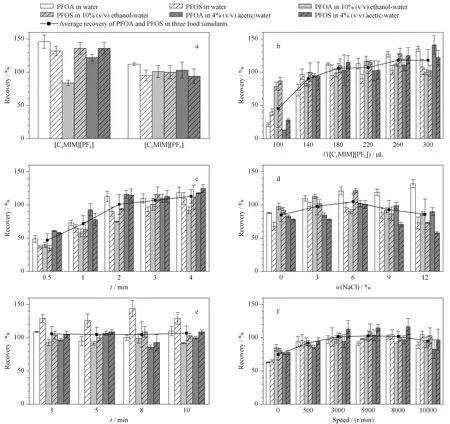
图 2 (a)萃取剂种类、(b)萃取剂用量、(c)涡旋时间、(d)氯化钠用量、(e)离心时间以及(f)离心速度对萃取效率的影响(n=3)Fig. 2 Effects of (a) extraction solvent types, (b) amounts of extraction solvent, (c) vortex time, (d) amounts of NaCl, (e) centrifugal time, and (f) centrifugal speed on the extraction efficiencies (n=3)
然后考察了萃取剂[C6MIM][PF6]的用量对萃取效率的影响,结果见图2b。将[C6MIM][PF6]体积分别设置为100、140、180、220、260和300 μL,可见随着用量增加,PFOA和PFOS的萃取回收率呈提高趋势。当[C6MIM][PF6]用量为220 μL时,3种食品模拟物中PFOA的萃取回收率为102.4% ~117.0% , PFOS的萃取回收率为90.8% ~115.2% ,并基本趋于稳定,故选取萃取剂最佳量为220 μL。
2.2 涡旋时间的优化
在离子液体分散液液微萃取中,分析物通过萃取剂和样品溶液间的充分接触可迅速达到分配平衡,但离子液体的高黏度特性阻碍了分散过程,降低了萃取效率,因此引入涡旋可促进疏水性的离子液体与食品模拟物中的目标分析物充分混合均匀,以便形成良好的乳浊液状态,促进传质过程。考察了涡旋时间对待测物萃取效果的影响,结果见图2c。随着涡旋时间的延长,3种食品模拟物中PFOA和PFOS的萃取回收率均呈先上升后稳定的趋势。在3 min时,PFOA和PFOS的萃取回收率分别为101.2% ~110.5%和90.2% ~116.3% 。4 min时,回收率无明显变化,因此,选择涡旋时间为3 min。
2.3 氯化钠质量浓度的优化
在分散液液微萃取中,加入无机盐可降低目标化合物在样品溶液中的溶解度,增加其在有机相的分配系数。本试验在样品溶液中加入5 mL不同质量分数(0~12% )的氯化钠评价无机盐对萃取效率的影响,结果如图2d所示,整体而言,在3种食品模拟物中,随着氯化钠用量的增加,PFOA和PFOS的萃取效率呈现先升高后下降的趋势。当氯化钠质量分数为6%时,[C6MIM][PF6]对PFOA和PFOS的萃取回收率分别为90.7% ~121.0%和96.8%~122.1% ,萃取效率最高。因此,氯化钠的质量浓度选择为6% 。
2.4 离心速度和离心时间的优化
离心是影响离子液体分散液液微萃取的一个重要步骤。离心温度高,会提高分析物以及离子液体在食品模拟物中的溶解度,不利于分析物的萃取,所以,本试验选择在4 ℃条件下进行离心。离心时间过短或者转速较低,均可能会使相分离不够充分,降低萃取效果。本试验选择在4 ℃条件下,考察不同离心时间和离心速度对萃取效果的影响(见图2e和2f)。结果表明,离心时间(3、5、8和10 min)对萃取回收率并没有显著影响;而在一定转速范围内,随离心速度的增大,萃取回收率提高,之后略有下降。当离心速度为5 000 r/min时,[C6MIM][PF6]对PFOA和PFOS的萃取回收率分别为91.5% ~104.0%和98.6% ~115.1% ,萃取效率基本达到最佳状态。因此,选择5 000 r/min离心3 min进行离心处理。
2.5 方法学考察
2.5.1线性范围、检出限以及定量限
在优化的实验条件下,将一系列不同质量浓度的空白基质加标溶液进样,以峰面积(y)对质量浓度(x, μg/L)进行线性回归,绘制标准工作曲线。试验结果表明,PFOA和PFOS在各自的线性范围内呈良好线性关系(r2>0.99)。分别以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)计算方法的检出限和定量限。PFOA和PFOS的线性方程、检出限及定量限见表2。PFOA在2~1 000 μg/L, PFOS在5~1 000 μg/L范围内线性关系良好(r2>0.99), 2种化合物的检出限分别为0.5和1 μg/L,定量限分别为2和5 μg/L。

表 2 PFOA和PFOS的线性方程、检出限及定量限Table 2 Linear equations, limits of detection (LODs), and limits of quantitation (LOQs) of PFOA and PFOS
y: peak area;x: mass concentration, μg/L.
2.5.2回收率与精密度
以不含待测物的食品接触材料样品为空白基质,分别在2~500 μg/L范围内添加低、中、高3个水平的混合标准溶液,按照上述优化的方法进行分析测定,每个水平平行操作6次,计算加标回收率。结果表明,3种食品模拟物中,PFOA和PFOS的平均回收率分别为86.4% ~115.3%和89.8% ~116.9% ,相对标准偏差为4.3% ~12.2%和4.6% ~14.4%(见表3),可满足实际检测需求。
2.6 实际样品分析
选取市面上销售的一次性纸杯、饮料瓶、酸奶瓶、桶装方便面等10件食品接触材料样品,根据食品接触材料样品预期接触的食品类别和属性,参照食品安全国家标准GB 31604.1-2015的规定,相应地从水、10%乙醇水溶液或4%乙酸水溶液3种食品模拟物选择一种适宜的测试介质,每个迁移试验平行制备3份试样,于70 ℃恒温水浴12 h,以本研究建立的优化方法对迁移液进行处理并测定PFOA和PFOS的迁移量。试验结果表明,2件食品接触材料在水模拟物中检测出PFOS,其迁移量分别为8.71和8.46 μg/L;1件食品接触材料在10%乙醇水溶液模拟物中检出PFOS,其迁移量为8.50 μg/L;其余食品接触材料的迁移液中均未检出PFOA和PFOS。
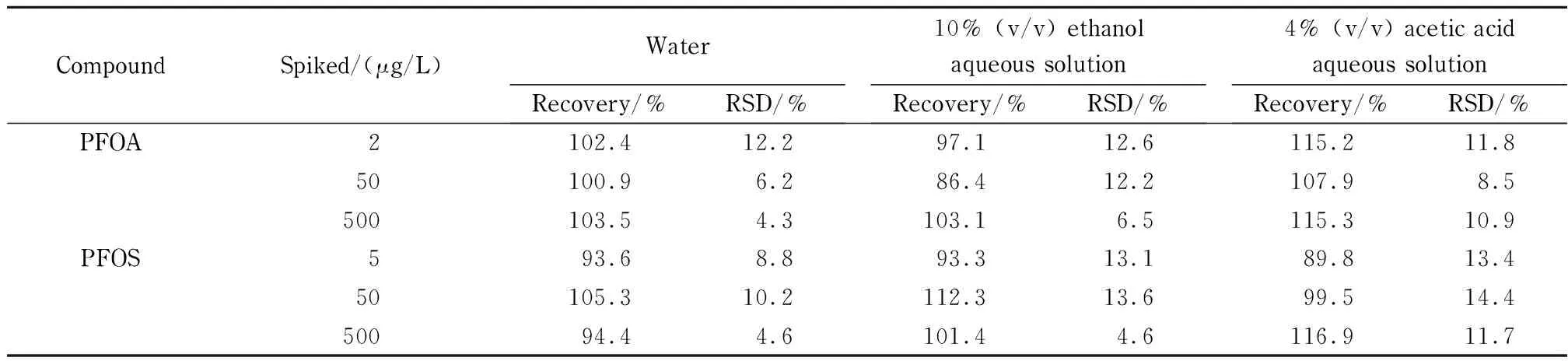
表 3 PFOA和PFOS的加标回收率和相对标准偏差(n=6)Table 3 Spiked recoveries and relative standard deviations (RSDs) of PFOA and PFOS (n=6)
3 结论
本研究建立了离子液体分散液液微萃取结合超高效液相色谱-串联质谱测定食品接触材料中PFOA和PFOS迁移量的分析方法。分别对离子液体种类和用量、涡旋时间、氯化钠质量浓度、离心时间及速度等因素进行了详细考察。该方法准确高效、环境友好,适用于食品接触材料中PFOA和PFOS迁移量的测定。
猜你喜欢
北京航空航天大学学报(2022年7期)2022-08-06
理化检验-化学分册(2020年5期)2020-06-15
分析化学(2019年3期)2019-03-30
中央民族大学学报(自然科学版)(2018年1期)2018-06-27
造纸化学品(2017年1期)2017-01-21
化工生产与技术(2016年5期)2016-03-13
化工生产与技术(2016年5期)2016-03-13
岩矿测试(2015年3期)2015-12-21
山东工业技术(2015年21期)2015-11-04
分析化学(2015年7期)2015-07-30

- 色谱的其它文章
- 全二维气相色谱-质谱分析煤油基吸热型碳氢燃料烃族组成
- 3种前处理方法处理婴幼儿配方奶粉中二十二碳六烯酸、花生四烯酸、亚油酸和α-亚麻酸的差异性
- QuEChERS-超高效液相色谱-串联质谱法测定不同植被类型土壤中11种甲氧基丙烯酸酯类杀菌剂
- 超高效液相色谱-串联质谱法快速测定鸡蛋中氟虫腈及其代谢物残留
- 固相萃取-高效液相色谱-串联质谱法同时测定牛奶和羊奶中莫奈太尔及其代谢产物残留
- Determination of 11 synthetic musks in imported seafood by solid phase extraction and gas chromatography-mass spectrometry