
Role of toll like-receptor 2 in inflammatory activity of macrophage infected with a recombinant BCG expressing the C-terminus of merozoite surface protein-1 of Plasmodium falciparum
2018-08-02 02:07NorMunirahZakariaRapeahSuppianNorazmiMohdNorNorFazilaCheMat
Nor Munirah Zakaria, Rapeah Suppian, Norazmi Mohd Nor, Nor Fazila Che Mat
School of Health Sciences, Health Campus, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
Keywords:Macrophages Mycobacterium bovis bacillus Calmette-Guerin Plasmodium falciparum Proinflammatory cytokine Toll-like receptor 2
ABSTRACT Objective: To investigate the role of toll-like receptor 2 (TLR2) in inflammatory activity of macrophage infected with the recombinant Mycobacterium bovis bacillus Calmette-Guerin(rBCG). Methods: Mouse macrophage cell line J774A.1 was infected with Mycobacterium bovis bacillus Calmette-Guerin (BCG) and rBCG cultures for 48 h in the presence or absence of 10 μg/mL of TLR2 inhibitor. Untreated macrophages were used as a negative control while lipopolysaccharide-stimulated macrophages were used as a positive control. The ability of the macrophage to engulf the BCG and rBCG in the absence or presence of TLR2 inhibitor was assessed using a phagocytic assay, while the production of inflammatory cytokines and nitric oxide by the infected macrophages was evaluated using ELISA and Griess reagent method,while the expression of the inducible nitric oxide synthase was determined using Western blot analysis. Results: The results showed that blocking TLR2 function reduced the phagocytic activity, nitric oxide production and proinflammatory cytokine secretion such as TNF-α,IL-1β and IL-12p40 as well as inducible nitric oxide synthase expression in the infected macrophages. These data showed the importance of TLR2 in the activation of macrophages following BCG and rBCG infections. Conclusions: Through exploring the immunological mechanism which underlies the protection conferred by the candidate vaccine, this study will improve our understanding of the vaccine candidate’s mechanism to protect the host from malaria infection.
1. Introduction
The complexity of malaria parasite’s life cycle encourages the development of an effective vaccine to eliminate the parasite from host blood.
Malaria caused byPlasmodium falciparum(P. falciparum) is one of the most severe diseases worldwide. It is estimated that 216 million malaria cases and 445 000 malaria deaths occurred in 2016. The majority of the victims are children under 5 years old[1].
The development of a recombinant vaccine using live bacteria such asMycobacterium bovisbacillus Calmette-Guerin (BCG) is one of the potential vaccine strategies for the development of new vaccines against pathogenic diseases including malaria parasites[2,3]. By using this strategy, the C-terminus of merozoites surface protein-1 (MSP-1C) ofP. falciparumwas engineered into the genome of a BCG clone to produce a recombinant BCG (rBCG) vaccine[4]. MSP-1C is a 19 kDa blood-stage antigen produced by proteolysis of 195 kDa MSP-1 protein. This protein is responsible for protective immunity against malaria blood-stage infection[5,6]. Previous studies demonstrated that the vaccine candidate was capable to stimulate a higher production of inflammatory activity in infected macrophages[7,8].
The recognition of BCG by macrophage is mediated by the pattern recognition receptor including toll-like receptors (TLRs)such as TLR2. This interaction triggers the activation of NF-κβ and mitogen activated protein kinases pathways that modulate the inflammatory gene transcription[9]. This will cause the secretion of pro-inflammatory cytokines.
Many studies have been conducted to evaluate the role of TLR2 in the early recognition of BCG by phagocytic cells such as macrophage[10-12]. However, no study proved that TLR2 is also important in the recognition of rBCG with the immune cell.Therefore, this research was conducted to investigate the involvement of TLR2 in the inflammatory activity of macrophage infected with our rBCG vaccine.
2. Materials and methods
2.1. Preparation of Mycobacterium bovis BCG and rBCG cultures
Mycobacterium bovisBCG was generously given by Professor Norazmi Mohd Nor from the School of Health Science, Universiti Sains Malaysia. The rBCG containing the MSP-1C ofP. falciparumwas constructed by Nurul (2007) using assembly polymerase chain reaction in the previous study[13]. BCG and rBCG cultures were grown in 7H11 agar supplemented with Middlebrook oleic acid,albumin fraction Ⅴ, dextrose and catalase (OADC) and in the addition of 15 μg/mL of kanamycin for rBCG. Both of the BCG and rBCG were incubated at 37 ℃ for 2 weeks. The bacteria were transferred to 7H9 broth supplemented with OADC for 1 week until the optical density of the culture reached ~0.8 at absorbance 600 (A600). Ten mL of the bacterial suspension was then centrifuged at 1 500 ×gat room temperature. The pellet was washed with PBS and then was resuspended in DMEM. The colony-forming unit of the bacteria was calculated using a spectrophotometer at absorbance 600 nm.
2.2. Preparation of J774A.1 macrophage cells
The J774A.1 mouse macrophage cell line (ATCC, USA) was cultured in complete DMEM culture medium. The cells were grown at 37 ℃ in a humidified incubator with the presence of 5% CO2.
2.3. Infection of J77A.1 macrophage with BCG and rBCG in the presence or absence of TLR2 inhibitor
Approximately 1 × 105macrophages were cultured overnight in a 96 well plate. After seeding overnight, the macrophages were inhibited with 10 μg/mL TLR2 inhibitor[14] (for the group in the presence of TLR2 inhibitor) for 30 min (based on optimization, data not shown)before both groups in the presence and absence of TLR2 inhibitor being infected with BCG and rBCG at multiplicity of infection of 1:20[7]. The cells were then incubated at 37 ℃ in the presence of 5% CO2for 48 h[8]. Uninfected cells were used as a negative control while cells stimulated with 100 ng/mL lipopolysaccharide (LPS)(Sigma-Aldrich, USA) were used as a positive control.
2.4. Cell viability assay
After 48 h, the media were replaced with 100 μL fresh DMEM. The assay was conducted in a 96 well tissue culture plate. Ten μL of filtersterilized stock 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Nacalai-Tesque, Japan) was added into each well of cell cultures and incubated at 37 ℃ for 4 h. One hundred μL of color development solution was then added and the plate was further incubated at 37 ℃ for an hour. Within an hour, the formazan crystals were dissolved and the absorbance intensity was measured by an ELISA plate reader at 570 nm with a reference wavelength of 630 nm. The cell viability assay was expressed as percentage of absorbance of the infected cells divided by the uninfected cells.
2.5. Phagocytic assay
The cell pellets were smeared with Ziehl-Neelsen Carbol-Fuchsin stain and heated for 5 min, after cell suspension was layered and fixed on a slide. Then, the cells were decolorized with 3% acid alcohol. After that, the cells were stained with methylene blue staining solution for 2 min before being washed with distilled water. The slides were air dried overnight before being mounted with distryrene plastieizer and xylene and examined under the light microscope at magnification × 100. The phagocytic index (PI) was calculated as the relative population of the mycobacteria in total of 100 macrophages.
2.6. Pro-inflammatory cytokine detection
The production of proinflammatory cytokines such as TNF-α, IL-1 β and IL-12p40 was determined in the culture supernatant of the cells using ELISA kit (Biolagend, UK). Briefly, the 96 well ELISA plate was coated with the specific antibody for IL-1β, TNF-α or IL-12p40 and incubated overnight at 4 ℃. The plate was then washed 5 times with PBS-T20. The pro-inflammatory cytokines productions of macrophages infected with BCG and rBCG in the presence and absence of TLR2 were done according to the manufacter’s instructions. Stop solution was added to stop the reaction and the plate was then read using an ELISA reader at 450 nm. The concentration of the pro-inflammatory cytokines produced by the cells was calculated by generating a standard curve.
2.7. Nitrite oxide (NO) detection assay
The NO production was determined in the supernatant of the infected and uninfected cells using Griess Reagent System (Promega,USA) according to the manufacturer’s instruction. Briefly, 50 μL standards and samples were added to a 96-well plate. Fifty μL suphanilamide solution was added and the plate was incubated for 10 min at room temperature. Fifty μL of N-1-napthylethylenediamine dihydrochloride (NED) solution was then added and the plate was further incubated for 10 min in the dark. The optimal density was determined at 540 nm using an ELISA plate reader. The concentration of NO was determined by generating a standard curve.
2.8. Inducible nitrite oxide synthase (iNOS) protein analysis
The macrophages were centrifuged for 5 min at 800 ×g. The pellet was resuspended with 100 μL high salt RIPA buffer and incubated at 4 ℃ overnight. The concentration of the cell lysate was estimated using NanoDrop UV-visible spectrophotometer. The protein sample was mixed with sample loading buffer and heated for 5 min. The protein was then separated using SDS-PAGE and the expression of iNOS protein was evaluated using rabbit anti-mouse iNOS antibody(BD, USA) diluted 1:1 000 with PBST-20 as a primary antibody and goat anti-mouse antibody conjugated to horseradish peroxidase(DAKO, Denmark) diluted 1:2 000 with PBST-20 as a secondary antibody. The membrane was then incubated with 200 μL of solution A and 200 μL of solution B of ECL Western blot detection reagent(Nacalai Tesque, Japan) for 30 s before being exposed using the chemiluminescence imaging system. The intensity density value(IDV) of iNOS expression and β-actin protein expression was measured using ImageJ software programme.
2.9. Statistical analysis
The statistical analyses were performed using the Statistical Package of Social Sciences (SPSS) software version 22. All the data obtained were from independent experiment (n=3) and presented as the mean ± standard error of the mean (SEM). Then, the data were analyzed by repeated measures analysis of variance (RM ANOVA).P<0.05 was considered statistically significant.
3. Results
3.1. Viability of J774A.1 macrophages infections
As shown in Figure 1, the viability of the macrophages was significantly decreased following infection with BCG and rBCG, as well as when stimulated with LPS either in the absence or presence of TLR2 inhibitor. However, the highest reduction was observed in cells infected with rBCG culture followed by the cells infected with BCG culture. Moreover, the results showed that the viability of all cells in the presence of TLR2 inhibitor significantly decreased compared with the infected cells in the absence of TLR2 inhibitor.

Figure 1. Effect of TLR2 inhibitor on viability of J774A.1 macrophages infected with BCG and rBCG cultures at 48 hours of incubation.
3.2. Phagocytic activity of J774A.1 macrophages infections
The phagocytic activity of macrophage infected with rBCG was demonstrated higher than those infected with BCG either in the absence or presence of TLR2 inhibitor. However, in both BCG and rBCG conditions, the presence of TLR2 inhibitor reduced the phagocytic activity of the infected macrophages more significantly than that in the absence of TLR2 inhibitor. Figure 2A shows the example of a microscopic image of a single macrophage that phagocytosed BCG and rBCG cells while Figure 2B represents the mean PI of 100 selected cells.

Figure 2. Effects of TLR2 inhibitor on phagocytic activity of J774A.1 macrophages infected with BCG and rBCG cultures at 48 hours of incubation.(A)Comparison of morphology of macrophage at 100 × magnification; (i)uninfected cell, (ii) infected cell with BCG culture and (iii) infected cell with rBCG culture. Mycobacterium staining appears as red. (B) Quantification of phagocytic activity of infected J774A.1 macrophages in 100 randomly selected cells. Data are expressed as the mean of phagocytic index (PI) ± SEM for three independent experiments. *P<0.05 is considered as significantly different compared to uninhibited cells.
3.3. Proinflammatory cytokines production by J774A.1 macrophages infections
Overall, rBCG infected macrophages produced the highest level of IL-1β followed by BCG infected macrophages and LPS stimulated macrophages either in the absence or presence of TLR2 inhibitor.The presence of TLR2 inhibitor reduced the production of IL-1β cytokine in all macrophages compared to the absence of the TLR 2 inhibitor in all samples (Figure 3A).
For TNF-α production, the highest TNF-α production was detected in the supernatant of LPS-stimulated macrophages. When the TNF-α production in BCG and rBCG infected macrophages was compared,rBCG-infected macrophages produced higher IL-1β level than those generated by BCG-infected macrophages. Similar to IL-1β, the presence of TLR2 inhibitor reduced the TNF-α production in all cells.However, a significant reduction was only observed in LPS stimulated macrophages and BCG-infected macrophages (Figure 3B).
The rBCG infected macrophages also produced the highest level of IL-12p40 in the supernatant of the cells followed by LPS stimulated macrophages and BCG-infected macrophages (Figure 3C). Similar to IL-1β and TNF-α production, the IL-12p40 production was demonstrated lower in macrophages treated with TLR2 inhibitor.However, significant differences in IL-12p40 production in the absence or presence of TLR2 inhibitor were only observed in rBCG infected macrophages and LPS stimulated macrophages.
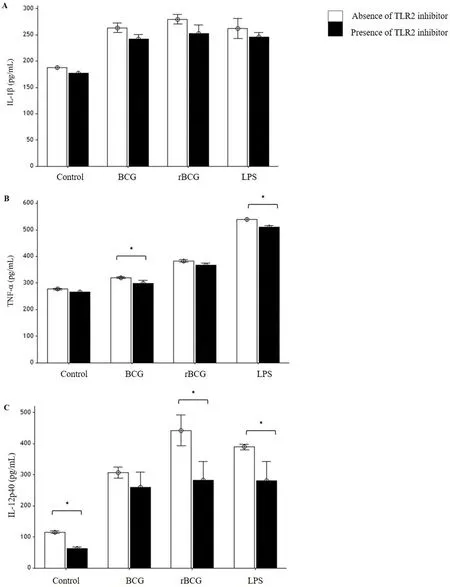
Figure 3. Effects of TLR2 inhibitor on IL-1β(A), TNF-α(B) and IL-12p40(C) productions by J774A.1 macrophages infected with BCG and rBCG cultures at 48 hours of incubation.
3.4. NO production and iNOS protein expression by J774A.1 macrophages infections
Different result was observed for iNOS protein expression.The highest iNOS expression was detected in rBCG infected macrophages (Figure 4A). However, the highest NO production was detected in macrophages stimulated with 100 ng/mL LPS either in the presence or absence of TLR2 inhibitor, followed by rBCG infected macrophages and BCG infected macrophages (Figure 4B). In line with other results, the presence of TLR2 inhibitor significantly reduced the NO production as well as iNOS expression in all macrophages.
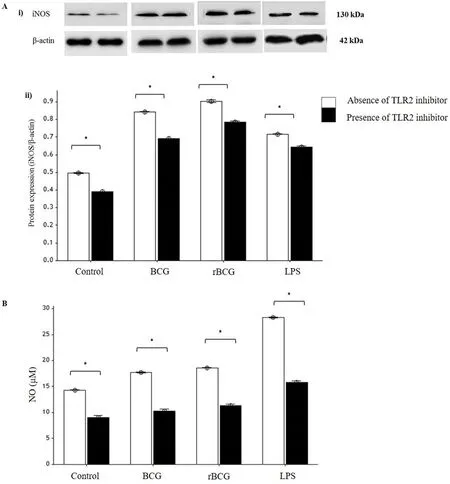
Figure 4. Effects of TLR2 inhibitor on iNOS protein expression (A) and NO production (B) by J774A.1 macrophages infected with BCG and rBCG cultures at 48 hours of incubation.
4. Discussion
TLRs are the crucial pattern recognition receptors involved in the initial interaction between the invading pathogens and macrophage[15].A lot of studies have suggested that TLR2 is involved in the initial recognition of mycobacteria such asMycobacterium bovisBCG by macrophage[14,16-19]. This interaction results in intracellular signalling cascades that leads to the activation of inflammatory activities such as pro-inflammatory cytokines and NO productions as well as iNOS expression[20-25]. This activation is mediated through the adaptor protein MyD88[26,27]. However, no study thus far has investigated the role of TLR2 in the recognition of rBCG by macrophage. Thus,this study was conducted to investigate the involvement of TLR2 in macrophage activation against the rBCG vaccine. Therefore, the involvement of TLR2 in the recognition of parent BCG and LPS was used as a comparison.
Our data demonstrated that the rBCG infection reduced the viability of infected macrophages considerably higher than BCG and LPS.This activity is important to destroy the invading pathogen in the phagolysosome of the phagocytic cell. This cellular mechanism is known as macrophage apoptosis. Our presented data are consistent with previous reports indicating thatMycobacterium bovisBCG strongly induced macrophage apoptosis in various cells such as THP-1. This was indicated by the reduced viability of the infecting BCG[28,29].Other studies also indicated that the surface engagement of TLR2 by mycobacteria could trigger apoptosis of the macrophages[30,31] which supported our data. The findings demonstrated that the inhibition of TLR2’s function increased the reduction of macrophage viability in both BCG and rBCG infected macrophages.
When BCG and rBCG clones were compared, the rBCG infected macrophages demonstrated a higher reduction in macrophage viability.We assumed that this is due to the presence of MSP-1C in the genome of the rBCG clone which increased the interaction of the rBCG with macrophage by stimulating more receptors such as TLRs. Both BCG and MSP1-C, capable of stimulating different or similar TLRs, thus stimulate higher macrophage activation than those produced by BCG alone. However, the receptor that mediates MSP-1C signalling and how the signal is transmitted into the macrophages have remained unclear. Thus far, no study demonstrated which TLR directly involved in the recognition of MSP-1C by macrophage. Data from previous study showed that proinflammatory responses to glycosylphosphatidylinositol of merozoite surface protein ofP. falciparumby macrophages are moderated through TLR2 and also highly extended through TLR4[22,32]. Since,glycosylphosphatidylinositol (GPI)[33] and MSPI-C are from the same organism and share similar characteristics, therefore we assumed that TLR2 might be also responsible for macrophage activation in response to MSP-1C.
The results also showed that rBCG infected macrophages generated higher levels of IL-1β, TNF-α and IL-12p40 compared to BCG infected macrophages. These data indicate that cloning the MSP-1C into the genome of the rBCG clone increased the interaction of the rBCG with macrophage, that led to the secretion of higher pro-inflammatory cytokine production. The data also showed that inhibition of TLR2’s function reduced the production of inflammatory cytokines by the cells.
NO, the free radical species produced by activated macrophage, mediates cytotoxic and cytostatic effects against pathogenic microbes[34,35]. In this study, the highest NO production was observed in macrophage following LPS stimulation. This data supported previous studies showing that NO synthesis in macrophage can be induced by microbial products such as LPS[36,37]. BCG and rBCG infections also led to increased NO production as indicated by higher nitric levels in culture supernatants of the infected macrophages although not as high as those produced by LPS stimulated macrophages. This result supported previous study showing that BCG is capable of stimulating macrophage activation due to the presence of lipoarabinomannan, a component of the BCG cell wall that is structurally related to LPS[38]. Although NO production by rBCG infected macrophage seems to be higher than NO production by the BCG infected macrophage, but this difference is not significant. The rBCG infected cells also produced higher iNOS expression and the expression was significantly reduced following treatment with TLR2 inhibitor. iNOS is the enzyme responsible for the conversion of arginine to NO. Once iNOS gene expression has been generated, macrophages produce large volumes of NO continuously[39].Raupach and Kaufmann suggested that cytokines such as IFN-γ and TNF-α stimulate macrophages to produce iNOS[40]. Therefore, we assumed that higher production of TNF-α in the culture supernatant of infected macrophages is responsible for the expression of iNOS in the infected cells.
In this study, we clearly indicate that TLR2 is activated in J774A.1 macrophage following infection with BCG and rBCG. This can be observed from the reductions of macrophage viability, phagocytic activity, pro-inflammatory cytokines productions, NO productions and iNOS protein expressions when the function of TLR2 in macrophages was inhibited. The ability of the rBCG to stimulate higher TLR2 function in macrophage which results in higher inflammatory activity of the phagocytic cells is important for the malaria parasite elimination.
These data also indicate that TLR2 is not the only TLR which is responsible for macrophage activation following BCG and rBCG infection. A number of studies revealed that TLR2 can also recognize a variety of microbial molecules by cooperating with other TLRs either as homodimer such as TLR2/2 or as heterodimer with TLR1 (TLR2/1) or TLR6 (TLR2/6)[41,42]. Indeed, previous study also suggested that, other TLRs such as TLR 4 also involved in the recognition of BCG and LPS by macrophages[43]. However,this phenomenon is not evaluated in this study. Therefore, the involvement of other TLRs should be studied in the future.
Conflict of interest statement
We declare that we do not have any conflict.
Acknowledgements
The research was supported by the Universiti Sains Malaysia(USM) Fundamental Research Grant Scheme (FRGS) (No. 203/PPSK/6171158).
 Asian Pacific Journal of Tropical Biomedicine2018年7期
Asian Pacific Journal of Tropical Biomedicine2018年7期
- Asian Pacific Journal of Tropical Biomedicine的其它文章
- Moderating effect of synthesized docosahexaenoic acid-enriched phosphatidylcholine on production of Th1 and Th2 cytokine in lipopolysaccharide-induced inflammation
- Characterization of Cnidoscolus quercifolius Pohl bark root extract and evaluation of cytotoxic effect on human tumor cell lines
- Anti-epileptic effect of morin against experimental pentylenetetrazol-induced seizures via modulating brain monoamines and oxidative stress
- Expression of fluorescent tagged recombinant erythroferrone protein
- Identification of a toxin coding fragment in pBSSB1, a linear plasmid from Salmonella enterica serovar Typhi that can stabilize a multicopy plasmid
- Influence of different cultivars of Phoenix dactylifera L-date fruits on blood clotting and wound healing