
一个LEOPARD综合征小家系临床表型及遗传学特征分析△
2018-07-28 08:58郭敏李书聆李锐洁江超武张力崔云华王芳朱兰阮标
听力学及言语疾病杂志 2018年4期
郭敏 李书聆 李锐洁 江超武 张力 崔云华 王芳 朱兰 阮标
在遗传性聋中,约22%为显性遗传,77%是 隐性遗传,1%是X连锁、线粒体突变母系遗传或Y连锁遗传[1]。根据是否有其他系统的疾病还可以分为综合征型聋和非综合征型聋,大约30%的遗传性聋为综合征型聋[2];常见的综合征型聋有Usher综合征、瓦登伯格综合征等。LEOPARD综合征是一类少见的常染色体显性遗传的综合征型聋,该综合征患者除表现为耳聋外,还可有皮肤、心脏等的异常,大约85%的LEOPARD综合征是由于PTPN11基因突变导致的[3]。本研究最近发现一个由PTPN11 c.1391G>C位点突变导致的LEOPARD综合征家系,现将该家系的临床表型特征及遗传学检查结果分析报告如下。
1 资料与方法
1.1研究对象 以先证者及家族成员为研究对象,先证者为女性,1岁4个月,因为未通过听力筛查来昆明医科大学第一附属医院耳鼻咽喉科进行听力诊断;其父母均为聋哑人,家系成员包括三代8名成员(图1)。本研究通过了本院伦理委员会批准,所有研究对象均签署了知情同意书,未成年人由监护人签署知情同意书。
1.2病史采集和全身体格检查 对先证者及家族成员进行完整病史(出生史、耳聋病史、家族史、耳毒性药物应用史等)采集和系统的全身体格检查。
1.3听力检测 对先证者进行听性脑干反应(ABR)、声导抗、耳声发射(OAE)检查,对家族其他成员进行纯音测听和声导抗检查。依据欧洲工作组倡导的遗传性聋分级标准进行耳聋程度分级[4]:轻度聋(20~40 dB HL)、中度聋(41~70 dB HL)、重度聋(71~95 dB HL)及极重度聋(>95 dB HL)。
1.4耳聋基因二代测序技术 抽取研究对象静脉血3 ml,用乙二胺四乙酸(EDTA)抗凝管保存,采用QIAamp DNA提取试剂盒(QIAGEN公司)抽提基因组DNA,对先证者进行已知的145个耳聋相关基因的外显子区进行靶向测序分析。这145个基因包含非综合征型耳聋基因和大部分综合征型耳聋基因(参见http://hereditaryhearingloss.org/)。所有数据用BWA算法比对到参考序列(UCSC hg19),采用仪器默认设置[5],对数据进行注释[6]。经筛选流程筛选,并结合患儿临床资料和生物信息学软件(Poly Phen2、Mutation Taster等)预测结果,对各个基因的功能、变异情况以及遗传模式进行分析,得到可疑候选突变,最后通过PCR扩增和Sanger测序对可疑候选突变的位点进行验证,并对患儿父母相应位点进行检测。PCR 扩增产物纯化与测序工作由广州金域检验所完成。选取听力正常对照组100例(健康体检人群)进行相应位点的检测,以排除遗传学上常见的无功能多态。
2 结果
2.1家系表型及全身体检结果 该家系表现为常染色体显性遗传,3代8人中,有3人表现为耳聋,分别为先证者(Ⅲ-1)、先证者父亲(Ⅱ-1)和母亲(Ⅱ-2),均为语前双耳重度感音神经性聋;其余家系成员均无耳聋病史。该家系的家系图见图1。
先证者及其父亲心前区可闻及杂音,超声心动图检查提示均有肥厚型心肌病。先证者父亲面部和颈部还可见到多发痣。其他家族成员全身无异常。
2.2听力检查结果 先证者听性脑干反应左右耳气导反应阈为90 dB nHL,双耳鼓室导抗图为A型,耳声发射不能引出;其父母双耳纯音听阈均>90 dB nHL,双耳鼓室导抗图均为A型。家族另一成员(Ⅱ-3)纯音听阈正常。
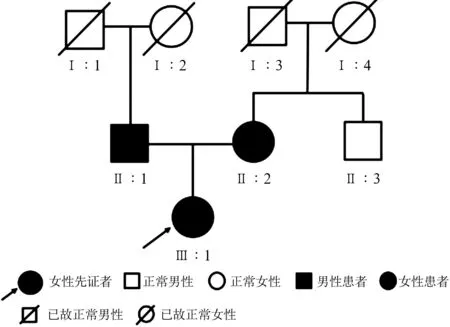
图1该家系常染色体显性遗传性聋家系图
2.3耳聋基因检测结果和生物学分析 通过二代测序,提示先证者及其父亲携带PTPN11基因c.1391G>C位点突变(图2);先证者母亲及对照组未发现该位点突变。使用Poly Phen-2 (http://genetics.bwh.harvard.edu/pph2/index) 和 Mutation Taster (http://www.mutationtaster.org/)进行突变对氨基酸功能影响的预测,提示该突变是一已知致病突变,会使编码蛋白的464位置的Gly 变成Ala (p. Gly 464 Ala),从而可能改变蛋白的功能;查询dbSNP, HGMD, 1000 Genomes Project, ClinVar and ESP6500数据库,未发现中国人有此突变的报道。

图2 先证者及其父亲PTPN11基因c.1391G>C突变测序峰图
3 讨论
LEOPARD综合征(LS, OMIM 151100) 是1936年由Zeisler and Becker 首次报道[3],该综合征的命名是由其临床特征的首字母组成:多发痣(multiple lentigines)、心电图传导异常(ECG conduction abnormalities)、 眼距过宽(ocular hypertelorism)、肺动脉狭窄(pulmonic stenosis)、 生殖器异常(abnormal genitalia)、 发育迟缓(retardation of growth)以及感音神经性聋(sensorineural deafness)。该综合征也曾被称为多发痣综合征(lentigines syndrome)、心脏皮肤综合征(cardio-cutaneous syndrome)、 莫尼汉综合征(Moynahan syndrome)等[7]。
LEOPARD综合征为常染色体显性遗传性疾病,可以是散发的,也可以是家族性的显性遗传,一旦父母一方患病,有50%的机率遗传给下一代。大约85%的LEOPARD综合征是由于PTPN11基因的错义突变导致的,该基因位于染色体12q24.1[8,9]。PTPN11基因负责编码SRC同源结构域的蛋白酪氨酸磷酸酶 (SHP2) 蛋白,该蛋白具有两个串联排列的SH2结构域(N-SH2和C-SH2)和一个蛋白酪氨酸磷酸酶(PTP)结构域;SHP2的功能是作为生长因子、细胞因子和激素的信号下调受体,并通过RAS-丝裂原活化蛋白激酶(MAPK)途径发挥特殊作用[3,10,11]。PTPN11突变可以导致LEOPARD 综合征和Noonan 综合征 (NS) (OMIM: 163950),50%的Noonan 综合征是由于PTPN11 突变引起[10]。虽然基因学相似,但是这两种综合征具有不同的表现和机制;LEOPARD 综合征患者基因突变是下调蛋白酪氨酸磷酸酶活性,而Noonan 综合征患者基因突变则会上调该酶的活性[12,13];相对于LEOPARD 综合征,Noonan 综合征更常见一些,其主要特征是身材矮小和面部畸形以及合并/不合并肥厚型心肌病[14,15]。
对于LEOPARD 综合征患者,心电图异常和进行性心肌传导异常是最常见的心脏疾病,通常肥厚型心肌病是威胁患者生命最常见的疾病[16~18]。多发痣是LEOPARD 综合征患者的一个特征表现,但是通常很少出现在5岁以下的患儿[8];LEOPARD 综合征患者的多发痣是扁平的、黑色至棕色的,主要分布在患者面部、颈部和上部躯干。感音神经性聋仅发生于15%~25%的LEOPARD 综合征患者[9,19],大多数患者都是出生后即出现耳聋,以后逐渐发展成为全聋。因此,对于LEOPARD 综合征患者来说,耳聋不像多发痣那样容易成为该疾病诊断线索。文中报道的这个家系中的先证者及患者就是以耳聋为首发症状,通过耳聋基因检测和家族成员的综合检查才得以确诊,从而进一步发现了患者及其家属的心脏和皮肤的异常。因此,详细了解耳聋患者的家族史和全身表现,同时对疑难病例进行基因检测,有助于正确的诊断和治疗。
目前研究显示PTPN11错义突变多位于7号、8号、12号和13号外显子,其中7号和12号外显子突变与肥厚型心肌病相关,8号外显子突变与肺动脉狭窄相关[20]。 对该家系的基因检测显示,先证者及其父亲是由于PTPN11 基因的c.1391G>C突变致病,该突变位点位于PTPN11 基因12号外显子,为一杂合错义突变;使用Poly Phen-2 和Mutation Taster 进行查询和功能预测,提示该突变是一已知致病突变,会使编码蛋白的464位置的Gly 变成Ala (p. Gly 464 Ala),从而可能改变蛋白的功能。通过查询ClinVar等数据库,该突变曾于2004年由Yoshida等首次报道[21],然而在中国人群中目前尚无此位点突变的报道,本研究结果丰富了我国综合征型聋的基因谱。
综上所述,本研究通过听力学检查显示听力异常,体检发现多发痣,超声心动图发现肥厚型心肌病,基因检测发现PTPN11 基因的c.1391G>C突变,明确了该耳聋家系的病因、临床表型和遗传特征,确诊先证者及其父亲为LEOPARD综合征,后续的干预措施为转诊患儿及其父亲到心内科门诊进行治疗和随访,并对该家系进行遗传指导,同时建议先证者进行人工耳蜗植入手术。
猜你喜欢
广西林业科学(2022年6期)2023-01-16
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中国生殖健康(2018年4期)2018-11-06
