
腘窝部未分化多形性肉瘤1例
2018-07-27 12:10范月莹王小兵
山西医科大学学报 2018年7期
范月莹,王小兵
(山西医科大学第一医院整形外科,太原 030001;*通讯作者,E-mail:zy-tina@163.com)
未分化多形性肉瘤(undifferentiated pleomorphic sarcoma,UPS)也被称为多形性未分化肉瘤或恶性纤维组织细胞瘤,是一种极少见的软组织恶性肿瘤[1]。现将我科收治的1例情况报告如下。
1 病例报告
患者,男,54岁,因“全身多发皮肤肿物50余年,左腘窝、下腹部肿物增大10年”就诊于山西医科大学第一医院整形外科。既往无特殊疾病史,病程中无发热等表现。
专科检查:患者全身浅表淋巴结未触及肿大,全身散在0.5 cm×0.5 cm×0.5 cm-2 cm×3 cm×3 cm大小不等的皮下结节,质软,无发红,无破溃,无压痛,腹背部及四肢皮肤广泛褐色色素斑,下腹部有一大小约11 cm×10 cm×6.5 cm的皮下肿物,左侧腘窝有一大小约14 cm×11 cm×10 cm的皮下肿物,表面凹凸不平,质软,无破溃,无压痛,无血管杂音,与基底无粘连,周围皮肤大致正常(见图1,2)。
入院诊断为:Ⅰ型神经纤维瘤病。
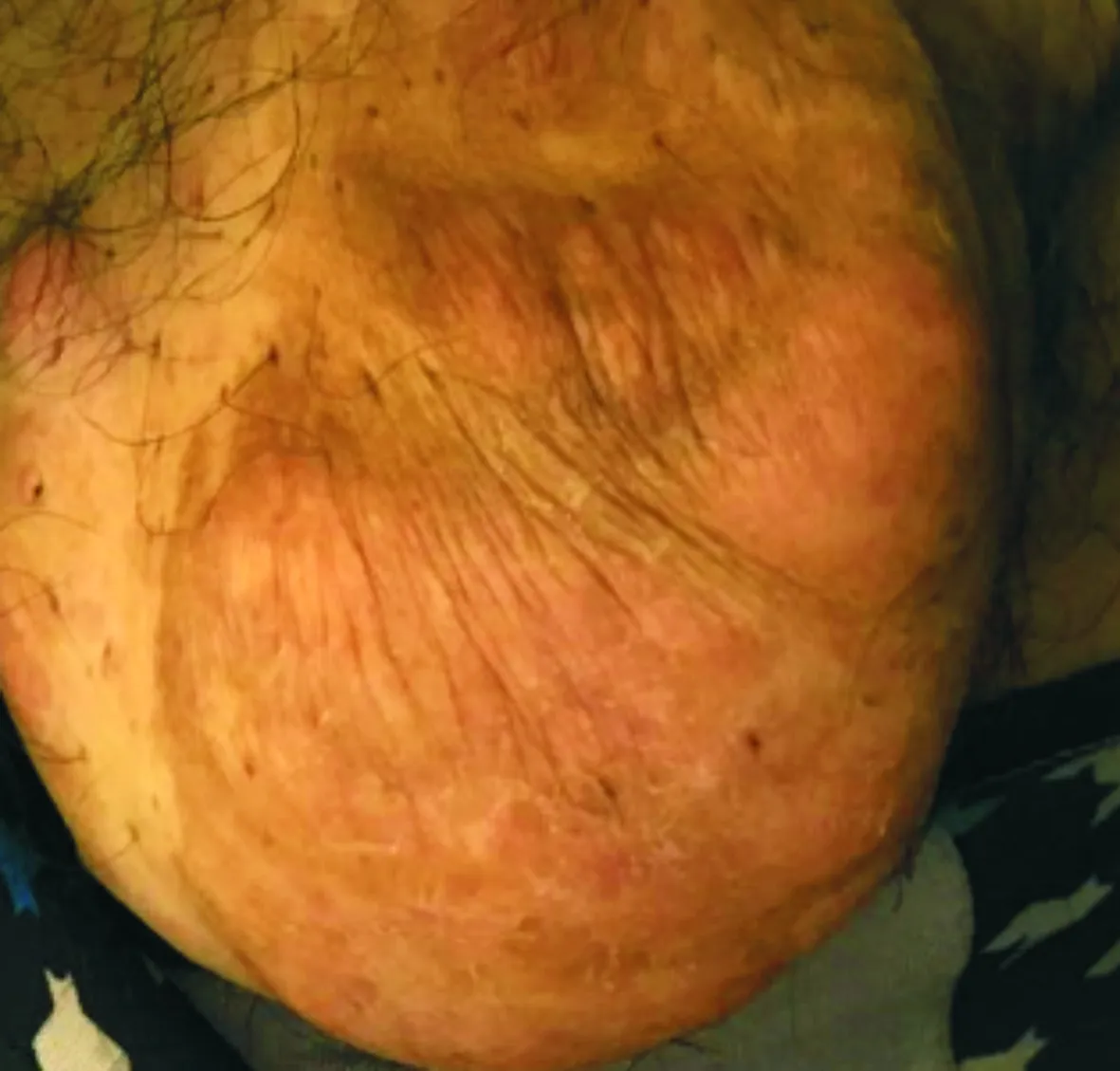
大小为11 cm×10 cm×6.5 cm图1 腹部巨大肿物
入院后完善相关化验检查,排除手术禁忌,于2017年12月15日在全麻下行腹部、左侧腘窝肿物切除缝合术+局部皮瓣转移修复术。依次切开皮肤、皮下,术中见下腹部类圆形肿物一个,于深筋膜表面彻底去除肿物,大小约11 cm×9 cm×6 cm,被覆皮肤,肿物底面与基底组织界限清楚,包膜完整,肿物剖开后,切面混杂,呈灰白、灰黄,实性,质中;术中见左腘窝部椭圆形肿物一个,于深筋膜表面彻底去除肿物,大小约14 cm×10 cm×9 cm,被覆皮肤,肿物底面与基底组织界限清楚,包膜完整,肿物剖开后,切面混杂,呈灰白、灰红,质中。
术后病理结果示:(腹部)神经纤维瘤,CD34(+)、CK(-)、Ki-67(-)、MBP(-)、S-100(+)、SMA(-)、Vimentin(+);(左侧腘窝)恶性梭形细胞肿瘤,Calponin(-)、CD34(血管+)、CD68(+)、CD99(-/+)、CEA(-)、CK(-)、EMA(-)、Ki-67(+10%)、MBP(-)、S-100(-)、SMA(血管+)、sox-10(-)、Vimentin(+)(见图3);北大医院会诊意见示:(左侧腘窝)多形性未分化肉瘤。

大小为14 cm×11 cm×10 cm图2 左腘窝部巨大肿物
最后诊断:①左侧腘窝多形性未分化肉瘤;②Ⅰ型神经纤维瘤病。术后切口愈合良好,恢复顺利,术后1个月复查左下肢功能恢复良好。患者及家属因经济原因,拒绝行化疗或放疗,出院3个月后复查,未见复发。
2 讨论
未分化多形性肉瘤也被称为多形性未分化肉瘤或恶性纤维组织细胞瘤,是一种极少见的软组织恶性肿瘤,曾被定义为兼有纤维母细胞和组织细胞分化的高级别梭形细胞恶性肿瘤,并认为是一特殊类型的肿瘤,是最常见的成年人软组织肉瘤[1]。在过去的10多年中,这种观点遭到争议和质疑。现在病理组织学普遍认为未分化多形性肉瘤是一种目前还不明确的组织细胞形态分化。未分化多形性肉瘤的病理诊断标准为:没有任何特异性分化方向的高级别肉瘤[2,3]。经严格评估后未分化多形性肉瘤占成人肉瘤不到5%[4]。UPS发病年龄高峰为50-70岁,大约2/3的UPS病例发生在男性,肿瘤最常发生在下肢,尤其是大腿,其次是上肢和后腹膜[5,6]。目前病因尚未明确,多认为与接触化学原料、放射、感染等多种因素有关。临床多表现为肿块、疼痛及压迫症状,早期可有血行转移,术后易复发等特点。目前认为软组织肉瘤来源于间质干细胞,能够解释软组织肉瘤的广泛分布。病理组织形态学检查是诊断的金标准[7]。影像学检查对诊断软组织肉瘤有辅助作用。MRI检查能够良好地显示软组织的对比度,因此为软组织肉瘤首选的检查方法,能够明确肿瘤的解剖位置、性质及其周围器官组织的关系,为手术方案提供重要的依据。尽管CT、MRI对其的检出率几乎达到100%,但是由于其影像表现多样,术前常被误诊或不能确诊。穿刺活检能够帮助术前明确诊断,但软组织肉瘤多存在不均质性,常需行免疫组化辅助诊断。

A.Calponin(-);B.血管内皮标记物CD34(+);C.CD68(+);D.CD99(-/+);E.EMA(-);F.Ki-67(+10%);G.S-100(-);H.平滑肌标记物SMA(+);I.Vimentin(+)图3 左侧腘窝组织免疫组化(DAB染色×100)
手术切除是首选治疗方法,根据肿瘤的部位、大小、分级、生物学特性多采用广泛切除或根治手术。广泛切除的手术原则是将肿瘤及肿瘤的活检部位1 cm内的正常组织完整切除并且获得阴性切缘[8]。对于肿瘤过大、位置较深而难以完整切除的,应根据肿瘤的类型及患者的综合情况考虑给予新辅助治疗(包括放疗及化疗)[7]。
神经纤维瘤病(neurofibromatosis,NF)又称多发性神经纤维瘤,是源于神经嵴细胞分化异常而导致的多系统损害的常染色体显性遗传疾病,常累及皮肤、神经、肌肉、骨骼和内脏,发病率低,半数患者有家族史,发病机制与等位基因突变与缺失、肿瘤微环境以及生长因子的作用有关[9]。1988年美国国立卫生研究院(national institute of health,NIH)将其分为神经纤维瘤病1型(neurofibromatosis type 1,NF1)和神经纤维瘤病2型(neurofibromatosis type2,NF 2)。其中NF1主要临床特征是多处皮肤咖啡色斑和多发神经纤维瘤。NF1的诊断标准为:①6个或以上的牛奶咖啡斑,青春期前最大直径5 mm以上,青春期后15 mm以上;②2个或以上任意类型神经纤维瘤或1个丛状神经纤维瘤;③腋窝或腹股沟褐色雀斑;④视神经胶质瘤;⑤2个或以上Lisch结节,即虹膜错构瘤;⑥明显的骨骼病变:如蝶骨发育不良,长管状骨皮质菲薄,伴有假关节形成;⑦一级亲属中有确诊NF1的患者。上述标准符合2条或以上者可诊断NF2[10]。本病病程长短不一,有时呈静止状态,可生存多年以至终生。本病例术后对标本进行病理检查及免疫组化示:(腹部)神经纤维瘤,符合诊断标准中①-③,临床诊断1型神经纤维瘤病成立。
目前国内外文献鲜有神经纤维瘤恶变为未分化多形性肉瘤的相关病例报道,而1型神经纤维瘤病合并局部巨大神经纤维瘤并恶变者未曾有报道。本病例中1型神经纤维瘤病与UPS是否具有同源性仍缺乏相关分子诊断的证据。
UPS各亚型具有相似的临床特征,当肿瘤发生在四肢时,表现为无痛性包块,持续时间为数月至数年。本病例在年龄、病史、发病部位均符合UPS诊断特征,然而该患者以神经纤维瘤病为主诊断入住我院,术前未给予特殊辅助检查及穿刺活检,手术肿物切除后,病理检查回报后得以确诊。神经纤维瘤病为常染色体显性遗传,多随年龄增长而恶变危险性增加,当神经纤维瘤瘤体短期内增大明显,破溃、出血及局部疼痛,均提示恶变可能,应及早诊治。因此,在目前的临床中,由于UPS发病率低,亦无特异性的临床表现及辅助检查手段及肿瘤标记物,外科医生又普遍缺乏对UPS的认知与重视,在临床上有较高的漏诊率及误诊率。近年来,全身各部位UPS的报道逐年增多,诊断多不能单纯依靠病史及辅助检查,往往需要更多的免疫组织化学的证据。因此,关于UPS尚需要新的诊断或预后分子标志物的研究,以及更多相关的临床研究及病例报道来探究未分化多形性肉瘤的细胞来源以及切实有效的治疗方案。
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
世界最新医学信息文摘(2021年12期)2021-06-09
幸福家庭(2021年1期)2021-03-08
中国临床医学影像杂志(2019年4期)2019-06-18
中国临床医学影像杂志(2019年4期)2019-06-18
中华老年口腔医学杂志(2016年3期)2017-01-15
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国卫生标准管理(2015年1期)2016-01-14
磁共振成像(2015年1期)2015-12-23
郑州大学学报(医学版)(2015年1期)2015-02-27
