
酱香大曲中链霉菌菌株FBKL4.005全基因组测序及序列分析
2018-07-24 08:45李豆南王晓丹罗小叶邱树毅
食品科学 2018年14期
李豆南,王晓丹,罗小叶,黄 魏,邱树毅,*
(1.贵州大学 贵州省发酵工程与生物制药重点实验室,酿酒与食品工程学院,贵州 贵阳 550025;2.贵州大学生命科学学院,贵州 贵阳 550025)
链霉菌(Streptomyces)从分类学上归属于放线菌目,是最为高等的放线菌类群,具有发育良好的分枝菌丝,主要分布于土壤环境中。研究表明,该类群主要是一类革兰氏阳性、化能异养型好氧菌[1],该菌属下的菌株多数能够合成结构复杂的次生代谢产物(如抗生素等),具有良好的生物活性,是一类具有很大应用潜力的微生物[2]。近年来,在酱香型、浓香型白酒酿造过程中也陆续分离得到该菌属下的多种菌株,并对其代谢特性进行了一定的探究,确认该菌属在酿造体系中存在重要的生物调控作用[3-4]。
全基因组测序是针对未知基因组序列的物种进行个体全部基因组测序,具有测序覆盖面广、准确性极高的技术优势[5]。近年来随着该技术的快速发展,以第2代测序技术为主体的全基因组测序技术已成为了一种高效检测手段,广泛应用于生物、医学等领域。在微生物学方面,以低错误率、低成本化和测序通量高为特征的罗氏454、Illumina HiSeq 2000测序平台已演变为基本的二代全基因组测序技术,成为了探究未知微生物类群多样性、生物学特性、代谢功能机制的重要手段[6-7],已经在土壤、肠道环境中微生物类群的分析鉴定以及工程菌株的开发中发挥了重要的作用[8-10]。针对一些极端环境中的微生物而言,由于其难以分离、可培养性低的特点,传统的分析方法已不适用,这时全基因组测序技术的优势便展现出来[11],其中,在诸如油田中嗜热采油芽孢杆菌(Geobacillus thermodenitrif i cans)[12]、新西兰地热区域中嗜酸甲烷氧化细菌[13-14]、水稻根际土壤中的斯氏假单胞菌(Pseudomonas stutzeri)[15]、蜡状芽孢杆菌[16]、深海沉积物中耐压耐冷的希瓦氏菌属[17]、两极地区永久冻土中的产甲烷菌类群[18]、氯消毒饮用水中的抗性变形菌类群[19]以及高酸性金属矿中的硫杆菌、酸微菌群落[20]等极端环境下微生物的研究中均有该技术的运用,为微生物防治石油污染、特殊代谢途径的发现和极端环境微生物多样性等方面的研究起到了巨大的推动作用。
本研究的开展建立在课题组王晓丹等[21]之前的研究之上,其利用罗氏454 FLX+高通量测序平台对贵州3 种酱香型高温大曲中细菌的群落结构多样性进行了深入研究,发现在属水平上链霉菌属为大曲中的优势细菌属之一,含量均占到大曲总生物量的1%以上,暗示该类群可能具有重要的功能性作用;基于这一结果,本课题组通过分离方法的设计,成功从大曲中得到1 株具有耐高温特性的链霉菌株FBKL4.005,并以该菌株为材料进行全基因组测序分析,从基因功能注释的角度快速剖析菌株的代谢特征,为日后酱香大曲中该类群功能性研究打下基础。
1 材料与方法
1.1 材料与试剂
链霉菌菌株(实验室编号:FBKL4.005)筛选分离自茅台某酒厂高温酱香大曲;细菌基因组DNA提取试剂盒 美国Biomiga公司;制霉菌素、新生霉素、琼脂糖、Tris、EDTA二钠 北京索莱宝科技有限公司;其他化学试剂均为国产分析纯。
1.2 仪器与设备
Thermo高速冷冻离心机 美国贝克曼库尔特有限公司;核酸凝胶电泳仪、凝胶成像仪 德国耶拿分析仪器股份公司;微型旋涡混合仪、Flex cycler多功能聚合酶链式反应(polymerase chain reaction,PCR)仪 美国Bio-Rad公司;BXM-30R立式灭菌锅 上海博讯实业有限公司医疗设备厂;SW-CJ-1D净化工作台 苏州净化设备有限公司。
1.3 方法
1.3.1 培养基的选择与配方
菌株纯化、斜面保藏培养基:ISP2培养基。
液体种子扩培培养基:葡萄糖10 g/L、酵母膏4 g/L、蛋白胨4 g/L、酵母浸粉4 g/L、K2HPO44 g/L、KH2PO42 g/L、MgSO4·7H2O 0.5 g/L,pH 7.2~7.4。
1.3.2 菌株总DNA的提取
从实验室低温(4 ℃)保藏的ISP2斜面培养基上刮取适量待测菌株,接种于100 mL液体种子扩培培养基中,于45 ℃培养24 h后,采用细菌基因组DNA提取试剂盒提取基因组DNA,操作步骤参照试剂盒说明书进行,所得基因组利用0.8%的琼脂糖电泳后在凝胶成像仪上对提取效果进行检测并用于基因组测序。
1.3.3 全基因组测序数据处理及分析
基因组DNA提取后采用Illumina HiSeq 2000测序平台完成全基因组测序,上机前先利用HiSeq平台完成建库,使用Agilent 2100对文库的插入片段进行检测,并运用实时定量PCR技术对文库的有效浓度进行准确定量,保证文库质量。将测序得到的原始数据进行过滤处理,分别去除质量值不大于38的低质量碱基、N碱基达到10 bp、与连接物之间overlap超过15 bp的reads,去除样品宿主以及重复污染,得到有效数据。
经过预处理后得到的有效数据,使用SOAP denovo组装软件[22-23]进行序列拼接组装,根据序列的双端信息,确定出contig排列,取长度大于500 bp的序列,根据不同的K-mer进行组装得到初步组装结果,然后采用krskgf、gapclose等软件对初步组装结果进行优化和补洞,将contig连接成基因组序列,得到最终组装结果,并与GO(Gene Ontology)、COG(Cluster of Orthologous Groups of proteins)、KEGG(Kyoto Encyclopedia of Genes and Genomes)、NR(Non-Redundant Protein Database)、TCDB(Transporter Classification Database)数据库进行比对分析,从而获得菌株基因组功能注释信息。
2 结果与分析
2.1 菌株基因组的组装
利用SOAP denovo组装软件[22-23]对测序得到的序列双端的信息数据进行拼接(原始序列67 771 429 个reads,读长150 bp,文库插入片段大小6 000 bp),共获得7 个scaffolds和69 个contigs,共9 441 858 bp的序列信息,其中用于判断基因组拼接效果优劣的N50大小为352 516 bp,N90大小为116 315 bp;经过优化与补洞处理后,获得菌株整个基因组大小为9 454 406 bp,GC含量为73.03%,基因平均长度为976 bp,其中编码区基因总数为8 316 个,占整个基因组序列大小86.01%。由菌株基因GC含量及测序深度分布图可知(图1),经过拼接组装后的GC含量主要分布在60%~80%之间,序列深度多分布在70~200×的范围;Unigene长度分布集中在300~1 100 bp及大于2 000 bp范围内(图2),其中400~500 bp长度的基因分布最多,达到669 个,占总数的8.00%左右。
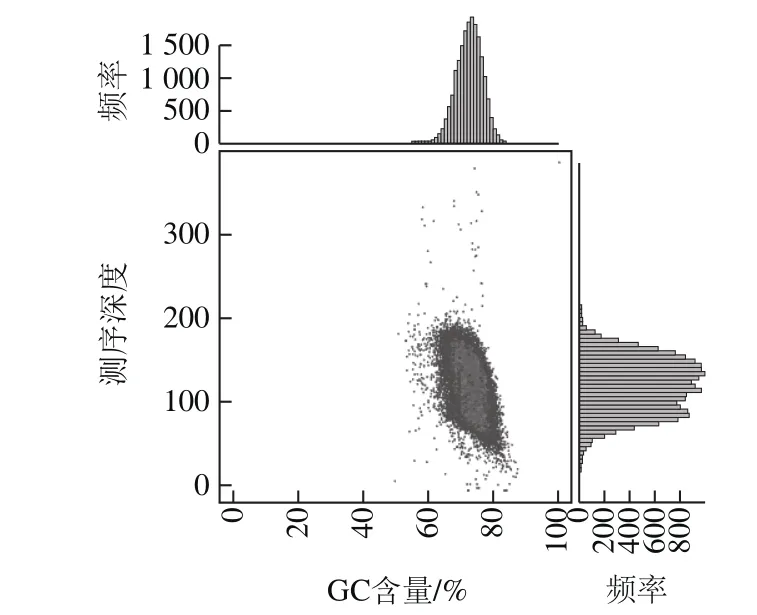
图1 菌株GC含量及测序深度分布图Fig.1 GC Content and sequencing depth distribution

图2 菌株Unigene长度分布Fig.2 Unigene length distribution
2.2 菌株基因组的功能注释
将菌株测序数据与GO(Released November,2013)、COG(Released September,2015)、KEGG(Released April,2016)、NR(Released April,2016)、TCDB(Released April,2016)功能数据库进行BLAST比对,同时使用antiSMASH(version 2.0.2)对基因组上已知次级代谢基因簇进行预测,对上述结果完成过滤(BLASTP,evalue≤1e-5),对于每一条序列的BLAST结果,均选取得分最高的比对结果(identity≥0%,coverage≥40%)进行注释。发现菌株基因组共有7 946 个基因得到成功比对注释,占基因总数的95.38%,共有258 个基因在所有数据库中得到注释,占基因总数的3.10%;其中,在NR、GO、COG数据库中得到功能注释的基因较多,分别为7 903、5 358 个和5 720 个,占基因总数的94.86%、64.31%及68.66%,而在TCDB数据库中得到注释的基因最少,仅有451 个,占基因总数的5.41%,具体的菌株基因组功能注释分布情况见图3。
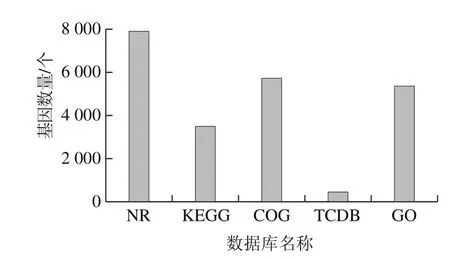
图3 菌株基因功能注释分布情况Fig.3 Database distribution of gene functional annotation from the strain
2.2.1 NR数据库基因注释分析
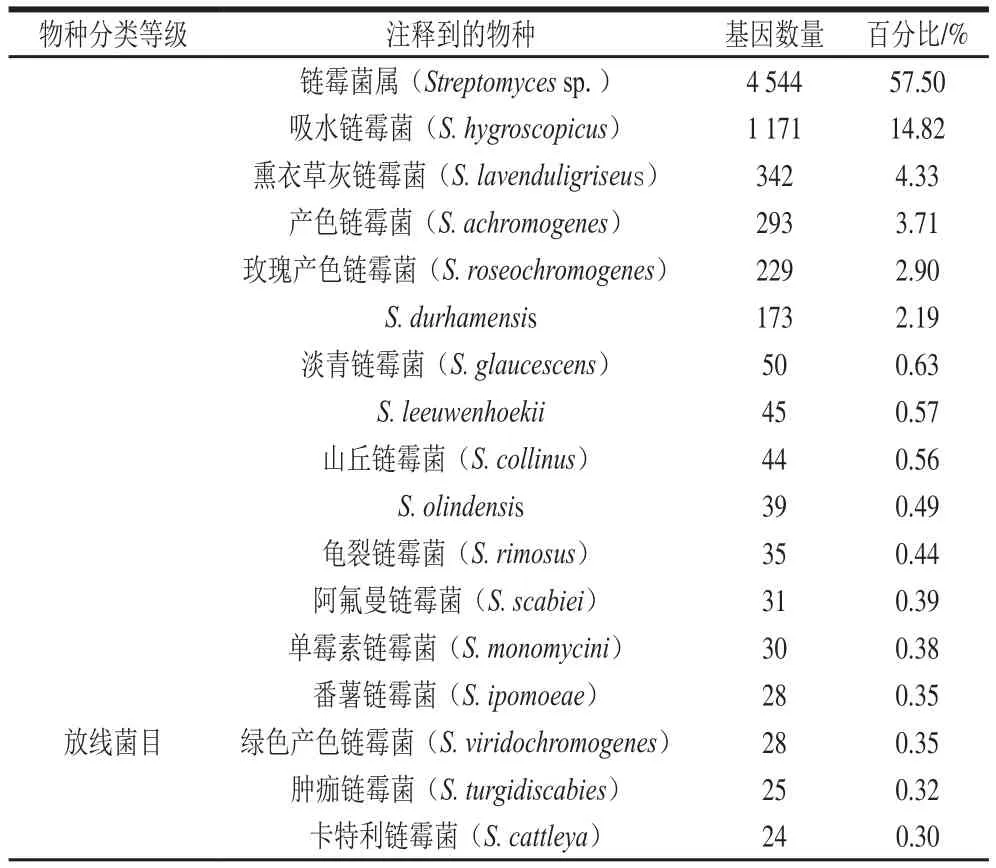
表1 菌株基因组NR数据库比对分析结果Table1 Results of BLAST with NR database

续表1
NR是一个非冗余的蛋白质数据库[24],通过BLAST软件,将菌株FBKL4.005的基因序列翻译为相应的氨基酸序列,并与NR数据库进行比对,得到最终的注释结果。菌株一共有7 903 个基因被注释到,占基因总数的94.86%,其中菌株FBKL4.005拼接基因组在属水平上与现有的链霉菌属(Streptomyces sp.)基因组核酸序列相似性在57.50%左右,总体相似度最高;而在种水平上则与吸水链霉菌(S. hygroscopicus)具有14.82%最高相似性。
2.2.2 COG数据库基因注释分析
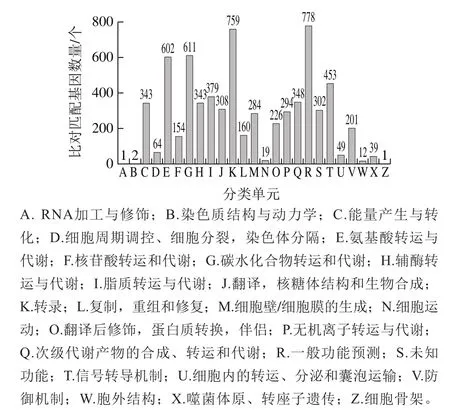
图4 菌株基因组COG数据库比对分析结果Fig.4 COG functional classif i cation of maitake Unigenes
COG是由NCBI创建并维护的蛋白数据库,根据细菌、藻类和真核生物完整基因组的编码蛋白系统进化关系分类构建而成[25]。利用BLAST软件,将菌株FBKL4.005的翻译得到的氨基酸序列,与COG数据库进行比对,完成相应基因的功能注释。从功能注释分析结果可以看出(图4),菌株FBKL4.005基因功能注释结果总共可以分为25 类,其中具有一般功能预测的注释结果最为丰富,共778 个,占注释基因总数的13.60%,其次是与物质代谢密切相关的759 个转录注释结果,占注释基因总数的13.27%,而与碳水化合物转运和代谢、氨基酸转运与代谢、信号转导机制等功能相关的基因也得到较多的注释结果,分别为611、602 个和453 个,此外还发现了302 个功能未知的基因,有待今后进一步研究。
2.2.3 GO数据库注释分析

表2 菌株基因组GO功能分类Table2 Gene ontology classif i cation of the strain
GO是1988年由基因本体联合会创立基因本体论数据库,能够通过细胞学组件、生物学途径、分子功能3 大分支数据库对物种基因组进行分类和准确描述[26]。将菌株FBKL4.005翻译氨基酸序列与GO数据库进行比对,对GO的不同分类比对结果进行统计,得到菌株功能基因含量的分布情况(图5)。菌株共有5 358 个基因在GO数据库中被注释到,在3 大类功能注释数据下又可分为43 种功能注释结果(表2),其中的细胞学组件类有10 个分支,共3 591 个基因注释结果,菌株基因组与细胞、细胞组分功能组表现出最高相关性,各有1 514 个;生物学途径类注释存在22 个分支,共10 615 个,其中菌株基因组与代谢过程、细胞过程、生物调节、生物过程调控等功能组相关性较高,分别为3 161、2 677、993 个和979 个;分子功能类有11 个分支共7 044 个相关性注释结果,其中菌株基因组与催化活性、连接功能组相关性最高,分别为2 987 个和2 697 个。

图5 菌株的功能基因含量分布情况Fig.5 Functional gene distribution of strain FBKL4.005
2.2.4 KEGG数据库注释分析
KEGG建立于1995年,是全面分析基因表达产物在细胞中的代谢途径及功能作用的重要参考,其中,最核心的便是KEGG PATHWAY数据库;通过数据库的比对查阅,可以快速方便地确定发挥某类功能相关的所有注释基因[27-28]。本研究将菌株的氨基酸序列与KEGG数据库进行比对,对KEGG不同分类比对结果完成统计,发现菌株基因组共有3 492 个基因得到注释,占菌株基因总数的43.95%,碳水化合物、氨基酸以及脂肪代谢为菌株基因组最主要涉及的几种代谢通路,分别有355、345 个和155 个基因注释结果。
进一步通过KEGG PATHWAY数据库分析,确定菌株有179 个物质代谢通路得到注释,其中嘌呤代谢(93 个)、糖酵解与糖异生作用(91 个)、丁酸甲酯代谢(82 个)、氨基苯甲酸酯降解代谢(79 个)、脂肪酸代谢(78 个)、精氨酸/脯氨酸代谢(76 个)以及氨基糖/核苷酸糖代谢(75 个)等通路与菌株基因组相比拥有较高的相关度(表3)。此外,通过比对结果的汇总还发现338 个与白酒特征风味物质产生通路、187 个与土腥味物质代谢通路、87 个与环境污染物降解通路、74 个与大分子糖类物质降解通路以及57 个与链霉素和新霉素等抗生素产生通路相关的基因;因此根据这些信息,初步推测该菌株在白酒酿造体系中可能与特征风味产生有一定的关系,同时可能还具有一定的抗生素代谢功能和大分子物质降解能力,为今后该菌株代谢功能的探究打下了基础。

表3 菌株基因组KEGG数据库主要代谢通路分析Table3 Main metabolic pathways of the strain from KEGG database
2.2.5 TCDB数据库注释分析
TCDB是转运蛋白分类数据库,包括离子通道的分类系统,该数据库提供了TC编号、描述信息和超过600 个转运蛋白家族的实例数据;而转移系统则以5 个级别进行分类,每个级别都对应于TC编号里的一个编号,用以表示特定类型的转运蛋白质[29]。研究中使用BLAST软件,将菌株的氨基酸序列与TCDB数据库进行比对,分别得到TCDB一级分类统计结果与二级分类统计结果。其中从一级分类统计结果来看(表4),菌株基因组涉及到初级主动运输以及电化学势驱动转运体功能的基因最多,分别为244 个与136 个,初步表明了菌株FBKL4.005可能多以主动运输、电化学转运的方式分泌和吸收各种物质;而从二级分类统计结果来看(图6),与单向传递体、协同转运子、逆向转运子转运以及P-P磷酸化驱动转运蛋白相关的基因最多,分别为211 个和136 个,为菌株主要的转运蛋白系统。

表4 菌株TCDB数据库一级分类统计结果Table4 First level classif i cation of the strain by TCDB database

图6 菌株TCDB数据库二级分类统计结果Fig.6 Second level classif i cation of the strain by TCDB database
2.2.6 菌株次级代谢基因簇分析
次级代谢产物是微生物在一定的生长时期,以初级代谢产物为前体合成的一系列大分子生长非必需物质;通过次级代谢基因簇注释分析可以看出(表5),菌株基因组有关萜烯类物质合成以及非核糖体肽合成酶的基因簇最多,各有5 个,此外还发现了与芳香类化合物合成有关的基因簇,其中萜烯类物质是酱香型白酒生产中链霉菌属的主要代谢产物,与酿造过程中的土霉味产生密切相关,同时在药香型以及浓香型白酒中也均有该类物质少量检出[30-32],是白酒中与风味形成有关的物质之一;而芳香类化合物则同样存在于各种香型的白酒中,同样是构成白酒风味的重要成分[33]。

表5 菌株次级代谢基因簇分析Table5 Secondary metabolic gene clusters of the strain
3 讨论与结论
本研究采用Illumina HiSeq第2代高通量测序技术对分离自酱香大曲的具有耐高温特性的链霉菌菌株FBKL4.005进行全基因组测序,经序列拼接组装后,确定菌株基因组大小为9 454 406 bp,是一种GC含量高达73.03%的微生物类群,同时将整合基因组数据与GO、COG、KEGG、NR、TCDB等几大基本数据库的进行比对分析,完成菌株基因组各方面功能的注释及数据统计工作,从分子生物学的角度探究了该菌株的生物学特性以及代谢功能机制。
总体而言,菌株FBKL4.005在基因上与链霉菌属尤其是该属下的吸水链霉菌具有最高的相似性,从功能预测的角度来看,COG数据库的比对结果显示转录、碳水化合物、氨基酸代谢是菌株主要的功能预测结果;通过GO数据库的比对,细胞、细胞组分、代谢过程、催化活性等注释结果呈现出较高的相关性;利用KEGG数据库中的代谢通路分析手段,确定了菌株主要代谢通路的组成,发现多个与白酒特征风味、土腥味、糖类物质降解、链霉素和新霉素产生等代谢通路相关的基因;此外,对菌株转运蛋白系统及次级代谢产物基因簇进行了探究,从结果来看转运子转运、磷酸化驱动转运蛋白系统主导的主动运输、电化学转运为菌株运输物质的主要方式,同时发现与白酒风味、土腥味物质代谢相关的基因结构。而针对类似菌株,荆新云[34]也完成了全测序分析,获得了全长8 047 771 bp的基因组、7 570 个编码基因以及23 个次生代谢产物基因簇,同时发现与其耐热性相关普遍胁迫蛋白基因的拷贝数比一般链霉菌高。与耐高温链霉菌4F相比,菌株FBKL4.005基因组更大、次生代谢基因簇较多,且发现了高温链霉菌4F所没有的风味代谢相关基因,但其耐热性基因却未能有效证实。
研究针对酱香大曲中具有耐高温特性的链霉菌菌株,采用全基因组测序技术对其生物学特性和代谢功能机制进行探究,得到大量基因组学信息,为今后深入开展酱香大曲中耐热性链霉菌代谢功能特征及相关机制的研究提供了重要的数据参考。
猜你喜欢
中国计量大学学报(2022年2期)2022-07-18
当代水产(2022年1期)2022-04-26
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17
中成药(2018年7期)2018-08-04
少儿科学周刊·少年版(2015年3期)2015-07-07
少儿科学周刊·少年版(2015年3期)2015-07-07
遗传(2015年5期)2015-02-04
中国肿瘤临床(2014年23期)2014-07-02
