
新型一步法制备环糊精有机聚合物整体柱及其应用
2018-07-23 06:12:58郭嘉亮汪锦才黄若诗林航张婷婷
暨南大学学报(自然科学与医学版) 2018年4期
郭嘉亮, 汪锦才, 黄若诗, 林航, 张婷婷
(1.佛山科学技术学院, 广东 佛山 528000; 2. 暨南大学 药学院, 广东 广州 510632)
由于独特的理化性质以及在分子识别能力上显示出无可比拟的优势,环糊精(cyclodextrin,CD)成为了众多色谱固定相单体中最为热门的研究对象之一[1-2].为了提高柱效,传统环糊精色谱柱的填料颗粒需要一再减小,引致其背压飙升,不仅不利于分离分析速度的提升,而且对仪器耐压设计也提出了更严苛的要求.近年来,整体柱(monolithic column)由于易改性、柱容量大、适用于快速分离分析等优势而备受关注.其中,由有机聚合物整体柱材料与环糊精单体结合而成的环糊精聚合物整体柱,在手性拆分[2]、样品富集[3]、固相萃取[4]、催化水解[5-6]等方面都发挥了重要作用,使得环糊精固定相得到崭新发展.
目前常见的环糊精聚合物毛细管整体柱,通常是通过先制备基质固定相,再进行表面修饰得到.这种“两步法”除了要求整体基质具有合适的孔径,还需预留可与功能化单体反应的活性基团,使之键合到整体材料表面,存在过程复杂、耗时费事、重现性不佳等缺点[7].还有一种制备方法,就是利用可聚合的环糊精单体,混合生孔剂、交联剂等,通过一步共聚合而直接制备整体柱.然而,这种“一步法”需含有可聚合双键基团的环糊精单体化合物,但通过化学修饰方式对环糊精引入可聚合双键存在很多困难,因而造成可选择的环糊精单体为数不多.此前,本课题组采用“点击化学”反应的方法,合成了含甲基丙烯酸官能团的β-环糊精衍生物单-6-(1H-1,2,3-三氮唑-4-甲基丙烯酸甲酯)-β-环糊精(PMA-BCD),然后以此为单体通过原位聚合反应一步法制备了新型的环糊精基质的聚合物整体柱 poly(PMA-BCD-co-EDMA)(图1,Column A)[8];但具体应用于手性拆分时,却难以获得良好结果.这可能是由于间隔臂(点击反应形成的刚性氮唑环)影响了环糊精自身的柔性及空腔结构造成.目前有关间隔臂对一步法制备的环糊精整体柱的影响仍鲜见报道.为证实这一猜想,本研究将合成具有间隔臂更长、柔性更佳的单体化合物单-6-甲基丙烯酰-乙二胺-6-脱氧-β-环糊精(MAC-EDA-BCD),以此为功能性单体,EDMA为交联剂,制备不同间隔臂的poly(MAC-EDA-BCD-co-EDMA)(图1,Column B),并进一步考察其色谱性能.

图1 整体柱的制备
1 材料与方法
1.1 试剂与耗材
甲基丙烯酰氯(methacryloyl chloride, MAC),单-6-乙二胺-6-脱氧-β-环糊精(mono-6-ethylenediamine-6-deoxy-β-CD, EDA-BCD)、乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate, EDMA)、偶氮二异丁腈(azodiisobutyronitrile, AIBN)、二甲基亚砜(dimethyl sulfoxide, DMSO)、异丙醇(isopropanol, IPA)、4-氯-扁桃酸、2-溴-扁桃酸、莨菪酸、舒洛芬、甲苯、硫脲(上海阿拉丁化学试剂有限公司),乙腈为色谱纯(默克化工).其他一般有机试剂购自广州化学试剂厂.石英毛细管(375 μm O.D.×100 μm I.D.)(河北永年锐沣),0.22 μm 滤膜(天津博纳艾杰尔科技有限公司).
1.2 仪器及设备
DF-101S集热式恒温加热搅拌器(河南予华仪器有限公司)、BSA223S 电子天平(德国Sartorius公司)、高压泵(美国 Haskel)、LX-0.12/10A空气压缩机(上海鲁辛实业有限公司)、ABI4000Q TRAP 三重四极杆质谱仪(美国 AB 公司)、Vario EL元素分析仪(德国 Elementar 公司)、Merlin 高分辨场发射扫描电子显微镜(德国Zeiss)、SPD-15C 紫外检测器(日本岛津)、DiNa-S 纳流泵(日本KYA)、Unimicro TrisepTM色谱工作站、Valco 四通阀(20 nL)、PB-10 pH 计(德国 Sartorius).
1.3 环糊精单体的制备
量取5.0 mL水与2.0 mL吡啶的混合液加入25 mL三颈圆底烧瓶内,降温至10 ℃,搅拌加入EDA-BCD(质量1.0 g,摩尔数0.85 mmol),搅拌溶解后,缓慢滴加MAC(质量0.14 g,摩尔数1.34 mmol).控温10~15 ℃搅拌反应2 h后,加入50.0 mL水,搅拌5 min后静置分液,有机相加入水萃取3次,每次30.0 mL,合并水相,加入乙酸乙酯洗涤3次(50.0 mL/次),收集水相,搅拌下将水溶液缓慢滴入400.0 mL丙酮中,洗出固体,过滤、收集滤饼,得淡黄色固体,50 ℃热风干燥6 h,收料得固体化合物单-6-甲基丙烯酸酯-乙二胺-6-脱氧-β-环糊精(MAC-EDA-BCD, 质量0.96 g, 质量分数84.21%).MS (ESI):m/z=1 246.1[M+H]+(图2).
图2 环糊精单体的合成
1.4 环糊精聚合物整体柱的制备
参考文献[1,7]报道预处理石英毛细管.将上述化学合成得到的MAC-EDA-BCD与交联剂EDMA,引发剂AIBN,按一定的比例在合适的生孔剂系统(双元生孔剂 DMSO/IPA)中超声混合形成均匀溶液.溶液灌满22 cm长度的预处理好的毛细管中,两端用橡胶塞密封,60 ℃恒温水浴反应12 h.另参考文献[8],通过一系列的条件考察和配比优化以后,后最终得到相应的poly(MAC-EDA-BCD-co-EDMA) 整体柱(图1,Column B).
1.5 整体柱的表征方法
参考本课题此前报道的方法[5-7],包括扫描电镜仪观察形态学微观结构、元素分析仪测定整体柱组成、微径液相法考察保留机理等,对石英毛细管进行表征.
1.6 色谱流动相的配制
所有流动相和溶剂均是色谱纯级别.流动相是由有机相(ACN)和水相(H2O或缓冲盐溶液)按照一定的比例混合均匀而成的,使用前需超声脱气处理.所用缓冲盐溶液,包括磷酸二氢钾(KH2PO4) 溶液和三乙胺乙酸酯(TEAAc)溶液,配制方法参考本课题组已发表的文献报道.所有分析样品一般溶解于溶剂ACN/H2O(VACN∶VH2O=85 ∶15),配制质量浓度为1 mg/mL,通过0.22 μm滤膜过滤备用.实验中所有样品的检测波长为214 nm.
1.7 分子对接
实验平台为美国Tripos公司 Sybyl 8.1 软件中的Sulflex-Dock模块,Linux 操作系统.除特别说明以外,所有参数均采用默认值.全部配体小分子的预处理以及对接时的设定均为同等条件.结合本课题组的相关经验,同时参考文献[9]报道方法,对接研究采用的 β-环糊精晶体结构下载于 PDB 数据库(PDB Code: 2VQ4),同时为减少其他影响,删除所有水分子及其他蛋白、配体分子,然后通过修改6-位上羟基为氮唑环及乙二胺来构建.手性配体4-溴扁桃酸小分子采用标准键长和键角,使用 Sketch Molecule 模块绘制,并采用 Tripos 力场分子力学程序 Minimize 进行结构优化,负载 Gasteiger-Hückel 电荷,以 Powel 能量梯度法进行能量优化,最大优化次数为2 000次,能量收敛标准为 0.001 kcal/mol,优化以确保结构的稳定性.通过Surflex-Dock程序对已经准备好的4-溴扁桃酸与 β-环糊精晶体结构进行对接,并为对接结果打分.设置口袋阈值(threshould)值为0.5,膨胀系数(bload)为 2,然后通过运算形成活性口袋,其他均为默认设置.Surflex-Dock 打分函数以 -lgKd为单位模拟结合能力,得分越高的构象被认为是最佳构象.
2 实验结果与讨论
2.1 整体柱的制备配比优化
参考Column A的制备经验[8],优化Column B的制备配比(表1).初步制备得到的整体柱B1,在毛细管内出现非常致密的透明胶体状结构;在B1的基础上调整生孔剂的质量比wDMSO/wIPA=75.0 ∶25.0调至70.0 ∶30.0(B2),B2形成的胶状结构中出现部分聚合物;B3在B2的基础上增加交联剂EDMA的比例,成胶情况得以解除,但是聚合结构过于松垮;调整单体与生孔剂的质量比从20.0 ∶80.0(B3)调至25.0 ∶75.0(B4),B4聚合结构偏致密,通透性不太理想;优化生孔剂质量比wDMSO/wIPA从70.0 ∶30.0(B4)调至65.0 ∶40.0(B5),最终得出B5聚合最均匀且通透性良好,因此采用该配比进行后续的研究.
表1整体柱的制备及优化
Table1Compositionsofthepolymerizationmixturesusedforthepreparationofmonolithiccolumnsandthecorrespondingpermeabilities

No.Monomer MixturePorogen SolventMonomer: PorogenMAC-EDA-β-CD/%EDMA/%DMSO/%IPA/%Monomer/%Porogen/%Permeability KB172.028.075.025.020.080.0GelB272.028.070.030.020.080.0GelB367.432.670.030.020.080.0Too slackB467.432.670.030.025.075.07.91 Mpa/(×10-14m2)B567.432.665.035.025.075.0GoodB667.432.667.532.525.075.03.32 Mpa/(×10-14m2)
2.2 整体柱的理化性质表征
通过扫描电子显微镜成像观察整体柱内聚合物的微观结构信息.毛细管整体柱的横切面可以直观反映原位聚合的情况以及材料的形貌(包括大孔、中孔或小孔结构).结果显示,整体柱在总体形态上呈现聚合良好的颗粒米状,聚合物与毛细管内壁紧密结合,同时聚合物骨架孔径分布比例恰当,未见坍塌情况,聚合微球均匀分布(图3).小孔保证了固定相比表面积,大孔则保证了整体柱的良好通透性能.
通过元素分析的结果考察Column B的化学组成.制备得到的整体材料C、O、H的质量分数分别为56.97%、35.45% 和6.95%.已知MAC-EDA-BCD的C、O、H的理论质量分数分别为46.30%、44.97% 和6.48%;EDMA的 C、O、H的理论质量分数分别为60.59%、32.29% 和7.12%.根据元素分析结果分析,聚合物中MAC-EDA-BCD与交联剂EDMA的摩尔比约为1∶3,与投料比基本符合,证明聚合情况符合预期;同时,环糊精质量分数也与Column A基本相当[8],适合用于下一步的对比评估.

图3 整体柱的扫描电镜图
通过微径液相色谱法表征整体柱的保留机理.以非极性化合物甲苯和极性化合物硫脲为测试化合物,乙腈、水为流动相.通过改变流动相中乙腈的质量分数,考察甲苯和硫脲的保留时间的变化(图4).当乙腈的质量分数低于80% 时,甲苯的保留时间随着乙腈质量分数的增加而显著降低,而硫脲的保留时间基本不变;乙腈质量分数大于80% 时,甲苯的保留时间基本不变,而硫脲的保留时间有所增长.由此可见,Column B的保留机理是一种包含亲水作用与反相作用的双重保留模式,这与Column A是一致的[8],说明两者的保留机理并没有显著差异.

图4 乙腈质量分数与保留时间的关系
2.3 整体柱的手性拆分效果
本实验采用4种手性药物及中间体作为评估对象,对Column A和Column B的手性拆分能力进行对比,考察不同间隔臂对β-环糊精整体柱手性分离能力的影响.从表2可以看到,Column B的手性分离效果明显优于Column A.以2-溴扁桃酸为例(图5),在分离条件一致的条件下,其在Column A和Column B整体柱上的分离度Rs分别为0.42和1.71.手性分离结果表明,相对于前期研究[8]得到的Column A,Column B整体柱手性分离能力更佳.
表24种手性化合物在ColumnA和ColumnB上的手性分离效果
Table2EnantioseparationsoffourchiralcompoundsontheColumnAandColumnB
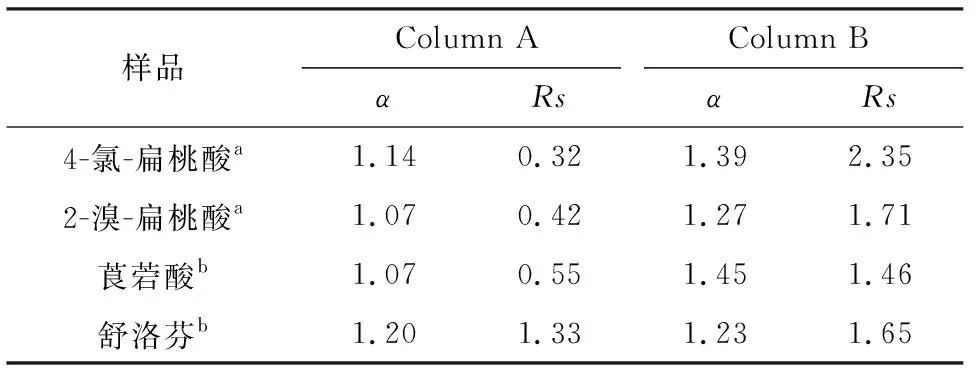
样品Column AColumn BαRsαRs4-氯-扁桃酸a1.140.321.392.352-溴-扁桃酸a1.070.421.271.71莨菪酸b1.070.551.451.46舒洛芬b1.201.331.231.65
流动相: (a)VACN∶VH2O=70∶30 containing 20 mmol/L KH2PO4, pH=4.20; (b)VACN∶VH2O=70 ∶30 containing 0.2% TEAAc, pH=4.20.
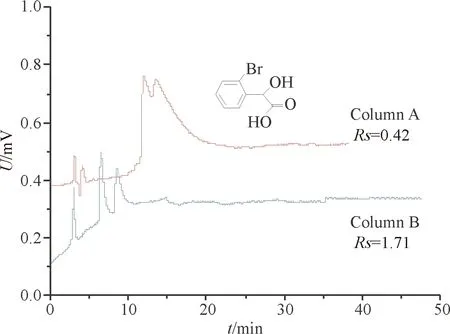
图5 4种手性药物在Column A 和 Column B 整体柱上的手性分离效果
Fig.5 Enantioseparation of 4 chiral drugs obtained on Column A and Column B
2.4 整体柱的手性识别机理的研究
利用分子对接手段,依据配体与受体作用的“锁-钥原理”(lock and key principle),有助于揭示 β-环糊精对对映异构体的拆分作用机理[9].通过对 PMA-BCD 和 EDA-BCD 与手性化合物 2-溴扁桃酸进行对接研究(图6).图6 a、b 分别显示为 S- 及 R-2-溴扁桃酸与 PMA-BCD 对接后的图示;图6 c、d 则分别显示为 S- 及 R-2-溴扁桃酸与 EDA-BCD 对接后的图示,可见 2-溴扁桃酸分子是以母环插入到 β-环糊精分子的疏水性空腔中,苯环的分子直径约为 5 Å,比 β-环糊精的内径 6.5 Å 略小,故通过苯环的插入形成包合物是合理的.并且由于苯环的插入,导致苯环上的手性羟基和羧基与环糊精大口侧的羟基空间距离靠近,形成强分子间氢键,使包合物更加稳定.因此,以此作为对接模型来研究 2-溴扁桃酸异构体的手性识别是合理的.
对接打分函数值如表3、表4所示.对 PMA-BCD 而言,最有代表性的 CScore 模块,所得分数相差无几,同为4;对 EDA-BCD 而言,R-2-溴扁桃酸与 S-2-溴扁桃酸的打分函数值为 2.740 0 和 2.730 0,但其他分数 (D_score、G_score、PMF_score、ChemScore、CScore)却存在着明显差别,最有代表性的 CScore 分别为1和4.从中发现,R-2-溴扁桃酸与 EDA-BCD的氢键作用显然要强于 S-2-溴扁桃酸,其分离主要原因可能是基于氢键作用力.
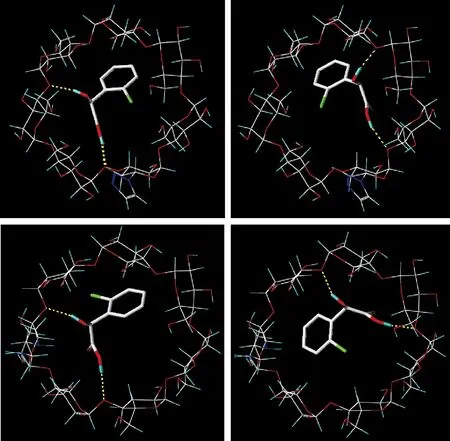
图6 2-溴扁桃酸不同对映异构体与 PMA-BCD (-S/-R(a)/(b)) 以及 EDA-BCD (-S/-R(c)/(d)) 分子对接的结果
Fig.6 The inclusion complexes of PMA-BCD and 2-bromo-mandelic acid racemate (-S/-R(a)/(b)) and the inclusion complexes of EDA-BCD and 2-bromo-mandelic acid racemate (-S/-R(c)/(d))

表3 2-溴扁桃酸2个对映异构体与 PMA-BCD 的6种分子对接分数Table 3 The Surflex-Dock scores of 2-bromo-mandelic racemates dock with PMA-BCD

表4 2-溴扁桃酸2个对映异构体与 EDA-BCD 的6种分子对接分数Table 4 The Surflex-Dock scores of 2-bromo-mandelic racemates dock with EDA-BCD
3 结论
本实验针对一步法制备环糊精有机聚合物整体柱进行深入探讨,以前期已有研究基础的PMA-BCD以及MAC-EDA-BCD为功能性配体,EDMA为交联剂,制得不同间隔臂的整体柱Column A 和 Column B,考察间隔臂对本法制备整体柱的影响.其中,Column B为首次报道.实验证明,Column B手性拆分效果优于Column A.影响化合物对映体在液相色谱中保留行为的因素很多,其中β-环糊精与对映体间结合物的稳定性差异是其主要因素[10-11].而其稳定性差异可在 Surflex-Dock 的结果 scores值上体现,即scores值越高,小分子与环糊精间相互作用越强,结合越牢固,形成结合物的稳定性越高.本实验中,R-2-溴扁桃酸与 S-2-溴扁桃酸的 CScore 差别显著,分别是4和1,R-2-溴扁桃酸形成的结合物更稳定,在色谱分离过程中后被洗脱,这与实验结果相吻合,表明分子对接有助于解释对映异构体的保留顺序.综上,Column A 和 Column B在手性分离效果上的差别原因可能由于前者单体间隔臂是刚性氮唑环,其空间位阻太大[8,10],间隔太短影响了环状柔性环糊精的空腔结构,而后者因为柔性的乙二胺连接方式,保证了环糊精足够的柔性,导致两者的差别.
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:30
中成药(2018年8期)2018-08-29 01:28:08
中成药(2018年6期)2018-07-11 03:01:20
中成药(2018年4期)2018-04-26 07:12:43
甘肃林业(2016年3期)2016-11-07 08:56:28
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:06
食品科学(2013年13期)2013-03-11 18:24:19
食品科学(2013年8期)2013-03-11 18:21:18
