
Selenocysteine antagonizes oxygen glucose deprivation-induced damage to hippocampal neurons
2018-07-23 02:58:58XianJunWangMeiHongWangXiaoTingFuYaJunHouWangChenDaChenTianSuYunBaiXiaoYanFu
中国神经再生研究(英文版) 2018年8期
Xian-Jun Wang , Mei-Hong Wang , Xiao-Ting Fu , Ya-Jun Hou Wang Chen Da-Chen Tian Su-Yun Bai Xiao-Yan Fu
Abstract Designing and/or searching for novel antioxidants against oxygen glucose deprivation (OGD)‐induced oxidative damage represents an effective strategy for the treatment of human ischemic stroke. Selenium is an essential trace element, which is bene ficial in the chemo‐prevention and chemotherapy of cerebral ischemic stroke. The underlying mechanisms for its therapeutic effects, however, are not well documented. Selenocysteine (SeC) is a selenium‐containing amino acid with neuroprotective potential. Studies have shown that SeC can reduce irradiation‐induced DNA apoptosis by reducing DNA damage. In this study, the in vitro protective potential and mechanism of action of SeC against OGD‐induced apoptosis and neurotoxicity were evaluated in HT22 mouse hippocampal neurons. We cultured HT22 cells in a glucose‐free medium containing 2 mM Na2S4O2, which formed an OGD environment, for 90 minutes. Findings from MTT, flow cytometry and TUNEL staining showed obvious cytotoxicity and apoptosis in HT22 cells in the OGD condition. The activation of Caspa se‐7 and Caspase‐9 further revealed that OGD‐induced apoptosis of HT22 cells was mainly achieved by triggering a mitochondrial‐medi‐ated pathway. Moreover, the OGD condition also induced serious DNA damage through the accumulation of reactive oxygen species and superoxide anions. However, SeC pre‐treatment for 6 hours effectively inhibited OGD‐induced cytotoxicity and apoptosis in HT22 cells by inhibiting reactive oxygen species‐mediated oxidative damage. Our findings provide evidence that SeC has the potential to suppress OGD‐induced oxidative damage and apoptosis in hippocampal neurons.
Key Words: selenium; selenocysteine; ischemic stroke; oxygen glucose deprivation; hippocampal neuron; mitochondria; reaction oxygen species;superoxide anion; oxidative damage; apoptosis
Introduction
Treating human stroke represents one of the biggest chal‐lenges in clinical practice. Approximately 80% of strokes are due to cerebral ischemia, which can lead to severe neu‐rological deficits and eventually death (Sveinsson et al.,2014; Mozaffarian et al., 2016; Wang et al., 2017a; Chang et al., 2018). The human brain is rich in polyunsaturated fatty acids, but contains low levels of endogenous antioxidant enzymes. Neurons are especially vulnerable to oxidative damage after cessation of blood flow to brain tissue (Juurlink and Sweeney, 1997; Li et al., 2007). Excess accumulation of reactive oxygen species (ROS) causes oxidative damage and can even lead to cell apoptosis (Fan et al., 2017a, b). Oxygen glucose deprivation (OGD) is the key condition underlying the pathogenesis of cerebral ischemia, resulting in oxidative damage and apoptosis. Therefore, designing and/or search‐ing for effective novel antioxidants against OGD‐mediated oxidative damage represents an effective strategy in the treatment of cerebral ischemia.
Selenium is an indispensable trace element, which is a fundamental component of selenoproteins and antioxidant enzymes in animals, including humans. It exhibits novel antioxidant properties by maintaining redox homeostasis(Fan et al., 2014). Considerable evidence indicates that tak‐ing a selenium supplement can effectively reduce the risk of human diseases (Gupta et al., 2003; Ozbal et al., 2008;Mehta et al., 2012; Fan et al., 2014; Pillai et al., 2014; Wang et al., 2016a, b; Zhao et al., 2017). Selenium inhibits the for‐mation of ROS, which can harm synapses, block cell com‐munication, and even result in neuronal apoptosis (Wang et al., 2012, 2016c; Singh et al., 2013). Selenocystine (SeC), a selenium‐containing amino acid, exhibits novel protective potential (Zhang et al., 2008; Chen and Wong, 2009). Kun‐war et al. reported that SeC can prevent γ‐radiation‐induced genotoxicity by reducing DNA damage (Kunwar et al.,2011). They also reported that 3,3′‐diselenodipropionic acid(DSePA), a SeC derivative, attenuates radiation‐induced DNA apoptosis by reducing DNA damage (Kunwar et al.,2010). Our previous study also showed that DSePA has the potential to suppress oxidative damage‐induced neurotoxic‐ity by inhibiting ROS generation and apoptosis in PC12 cells(Wang et al., 2016c). However, whether and how SeC might antagonize OGD‐induced neurotoxicity in HT22 mouse hippocampal neurons has not been investigated. Therefore,in the present study, we evaluated the protective potential and mechanisms of action of SeC against OGD‐induced neurotoxicity in anin vitrocell model.
Materials and Methods
Chemicals
SeC, sodium hydrosulfite (Na2S4O2), MTT, dimethyl‐sulfoxide (DMSO), propidium iodide (PI) and all other reagents were bought from Sigma (St. Louis, MO, USA).TUNEL‐DAPI kit, 2′,7′‐dichlorofluorescein diacetate(DCFH‐DA), MitoSOX, and BCA kit were purchased from Beyotime (Shanghai, China). Caspase substrates for caspase‐3, ‐8 and ‐9 and all antibodies were obtained from Cell Signaling Technology Inc. (Beverly, MA, USA).
Cell culture and drug treatment
HT22 mouse hippocampal neurons were bought from KeyGen Biotech (Nanjing, China) and were cultured with Dulbecco’s modified Eagle medium‐Ham’s nutrient mix‐ture F‐12 (DMEM‐F12) with 10% fetal bovine serum (FBS;Invitrogen, Carlsbad, CA, USA) at 37°C and 5% CO2. Glu‐cose deprivation was achieved using a glucose‐free Earle’s solution (Invitrogen). Experiments were carried out on four groups: the control group (untreated cells), the OGD group(104 cells in 96‐well plates treated with glucose‐free medium+ 2 mM Na2S4O2for 90 minutes), the SeC‐treated group (cells treated with SeC for 7.5 hours) and the protective group (cells pre‐treated with SeC for 6 hours, then treated with OGD for 90 minutes). Subsequently, the MTT method was used to detect cell viability, as described previously (Fan et al., 2014).
Flow cytometry analysis
After measuring cell viability, cell apoptosis was assessed using flow cytometry (BD Biosciences, Bedford, MA, USA)(Fan et al., 2017a). HT22 cells were incubated with 0.4 or 0.6 μM SeC for 6 hours and/or cultured under OGD (glu‐cose‐free medium + 2 mM Na2S4O2) for 90 minutes. After treatment, cells were harvested, fixed and loaded with PI.The Sub‐G1 peak was used to quantify the number of apop‐totic cells. Cell cycle distribution was analyzed using ModFit software (BD Biosciences). About 1 × 104cells per sample were recorded. Optical density was used to measure cell via‐bility, expressed as a percentage of control values.
TUNEL-DAPI assay
After flow cytometry analysis, OGD‐induced apoptosis in HT22 cells was measured using the TUNEL‐DAPI kit (Be‐yotime), as previously described (Fan et al., 2017a). Brie fly,cells were pre‐treated with 0.6 μM SeC for 6 hours and/or cultured under OGD (glucose‐free medium + 2 mM Na‐2S4O2) for 90 minutes. Then, cells were fixed, permeabilized,and incubated with TUNEL working solution and DAPI solution, respectively. After washing, cells were imaged un‐der an Eclipse80i fluorescence microscope (magnification,200×; Nikon, Tokyo, Japan). The number of TUNEL‐posi‐tive cells was manually counted and expressed as a percent‐age of the control count.
Evaluation of caspase activation
After quantifying cell apoptosis using flow cytometry and the TUNEL‐DAPI assay, caspase activation was measured using specific caspase substrates (Ac‐DEVD‐AMC for caspase‐3, Ac‐IETD‐AMC for caspase‐8, and Ac‐LEHD‐AMC for caspase‐9; all obtained from CST). HT22 cells were pre‐treated with 0.4 and 0.6 μM SeC for 6 hours and/or cul‐tured under OGD (glucose‐free medium + 2 mM Na2S4O2)for 90 minutes. Cells were then collected and total protein was prepared. 100 μg of total protein per well was incubated with caspase substrates at 37°C for 2 hours. Caspase activa‐tion was determined using a microreader, as described pre‐viously (Fan et al., 2014). These procedures were performed at least three times, and the mean caspase activation was expressed as a percentage of control values.
Detection of ROS and superoxide anions
After measuring cell apoptosis, the intracellular oxidative status of HT22 cells was evaluated using specific fluores‐cent substrates. Briefly, cells were pre‐labeled with 10 μM DCFH‐DA or 10 μM MitoSOX. Then, cells were washed and pre‐treated with 0.6 μM SeC for 6 hours and/or cultured under OGD (glucose‐free medium + 2 mM Na2S4O2) for 90 minutes. The production of ROS and superoxide anions was then quanti fied under a fluorescence microscope (Olympus,Tokyo, Japan).
Western blot assay
After assessing cell viability and apoptosis, the underlying mechanisms were examined by western blotting. Cells were pre‐treated with 0.6 μM SeC for 6 hours or/and cultured under OGD (glucose‐free medium + 2 mM Na2S4O2) for 90 minutes. Total protein was prepared and 40 μg was loaded per lane for separation by sodium dodecyl sulfate polyacryl‐amide gel electrophoresis (SDS‐PAGE). Protein was then transferred from the blot onto a nitrocellulose membrane,blocked and incubated overnight with anti‐rabbit mono‐clonal primary antibodies (1:1000; CST) at 4°C. Primary antibodies included cleaved‐PARP, active‐caspase‐3, ac‐tive‐caspase‐7, active‐caspase‐9, Ser1981‐ATM, Ser428‐ATR, Ser15‐p53, total‐p53 and Ser139‐H2A (all obtained from CST). The membrane was then incubated with goat‐anti monoclonal second antibodies IgG (1:2000; CST),at 37°C for 2 hours. The target protein of interest was im‐aged on X‐ray films using an enhanced chemiluminescence system (Kodak, Japan). β‐actin was used as the reference band. Protein expression (relative optical density) was quan‐ti fied by Quantity‐One Software (Bio‐Rad, CA, USA) based on the optical density of β‐actin.
Statistical analysis
All data and images were obtained from at least three exper‐iments and mean values were used for all statistical analyses.SPSS 17.0 software (SPSS, Chicago, IL, USA) was used to perform the statistical analysis. Data shown are presented as the mean ± SD. Intergroup comparisons were performed using a one‐way analysis of variance, followed by the least signi ficant difference test with a signi ficance level ofα= 0.05.
Results
SeC alleviates OGD-induced cytotoxicity in HT22 cells
As shown in Figure 1A, SeC (0.2–1.0 μM) treatment alone was not toxic to HT22 cells. In contrast, cells cultured under OGD conditions (glucose‐free medium + 2 mM Na2S4O2) showed a time‐dependent decline in cell viability(Figure 1B). For instance, cells cultured under OGD for 60,90 and 120 minutes signi ficantly decreased in cell viability to 75.3%, 64.2% and 51.8%, respectively. However, SeC pre‐treatment markedly alleviated OGD‐induced cytotoxic‐ity in HT22 cells. Pre‐treatment of cells with 0.4 and 0.6 μM SeC for 6 hours (Figure 1C) signi ficantly increased survival in OGD‐treated cells from 68.5% (treated with Na2S4O2) to 76.9% and 89.4%, respectively. Moreover, SeC pre‐treat‐ment also improved measures of cell morphological change.As shown in Figure 1D, SeC effectively reduced cell shrink‐age, increased cell numbers and increased cell‐to‐cell con‐tact in HT22 cells under the OGD condition. These results demonstrate that SeC alleviates OGD‐induced cytotoxicityin vitro.
SeC suppresses OGD-induced apoptosis in HT22 cells
The OGD‐induced cell death model system was explored using flow cytometry analysis. As shown in Figure 2A,cells cultured under OGD conditions showed significant cell apoptosis, as revealed by the increase of the Sub‐G1 peak. However, SeC pre‐treatment dramatically suppressed OGD‐induced apoptosis. Pre‐treatment of cells with 0.4 and 0.6 μM SeC for 6 hours significantly suppressed cell apoptosis from 34.4% (OGD condition) to 23.8% and 10.2%,respectively. SeC treatment alone did not differ in levels of apoptosis in HT22 cells compared with control. Statis‐tical analysis of the Sub‐G1 peak (quantifying apoptotic cells) confirmed this protective effect (Figure 2B). There was no significant effect on cell cycle distribution (Figure 2C). Furthermore, TUNEL‐DAPI assay results suggested that cells cultured under OGD exhibited obvious HT22 cell apoptosis, as indicated by the increased green fluorescence of TUNEL‐positive cells (Figure 2D, E). As expected, SeC pre‐treatment signi ficantly reduced apoptosis in HT22 cells(Figure 2D, E). Taken together, both of the cell viability methods used indicate that SeC has the potential to antago‐nize OGD‐induced HT22 cell apoptosis.
Role of mitochondrial-mediated apoptosis
Apoptosis can be triggered by mitochondrial (intrinsic)or death receptor (extrinsic) pathways, which are widely accepted as the two most important cell death pathways(Yagami et al., 2014). To characterize the speci fic cell death mechanism induced by OGD, the status of intracellular caspase activation was evaluated with three speci fic caspase substrates. As shown in Figure 3A, cells cultured under OGD conditions (glucose‐free medium + 2 mM Na2S4O2)showed signi ficant activation of caspase‐3, ‐8 and ‐9, indi‐cating that OGD triggered mitochondrial and death receptor apoptosis pathways. Caspase‐9, the main initiator of the mi‐tochondrial‐mediated cell death signal, was more active than caspase‐8 under OGD conditions. However, SeC pre‐treat‐ment significantly inhibited OGD‐induced caspase activa‐tion. Western blot results further con firmed this protective effect. As shown in Figure 3B, cells cultured under OGD showed obvious PARP cleavage and activation of caspase.SeC pre‐treatment reduced OGD‐induced PARP cleavage and caspase activation. Taken together, these results show that SeC has the potential to antagonize mitochondrial‐me‐diated apoptosis in OGD‐treated cells.
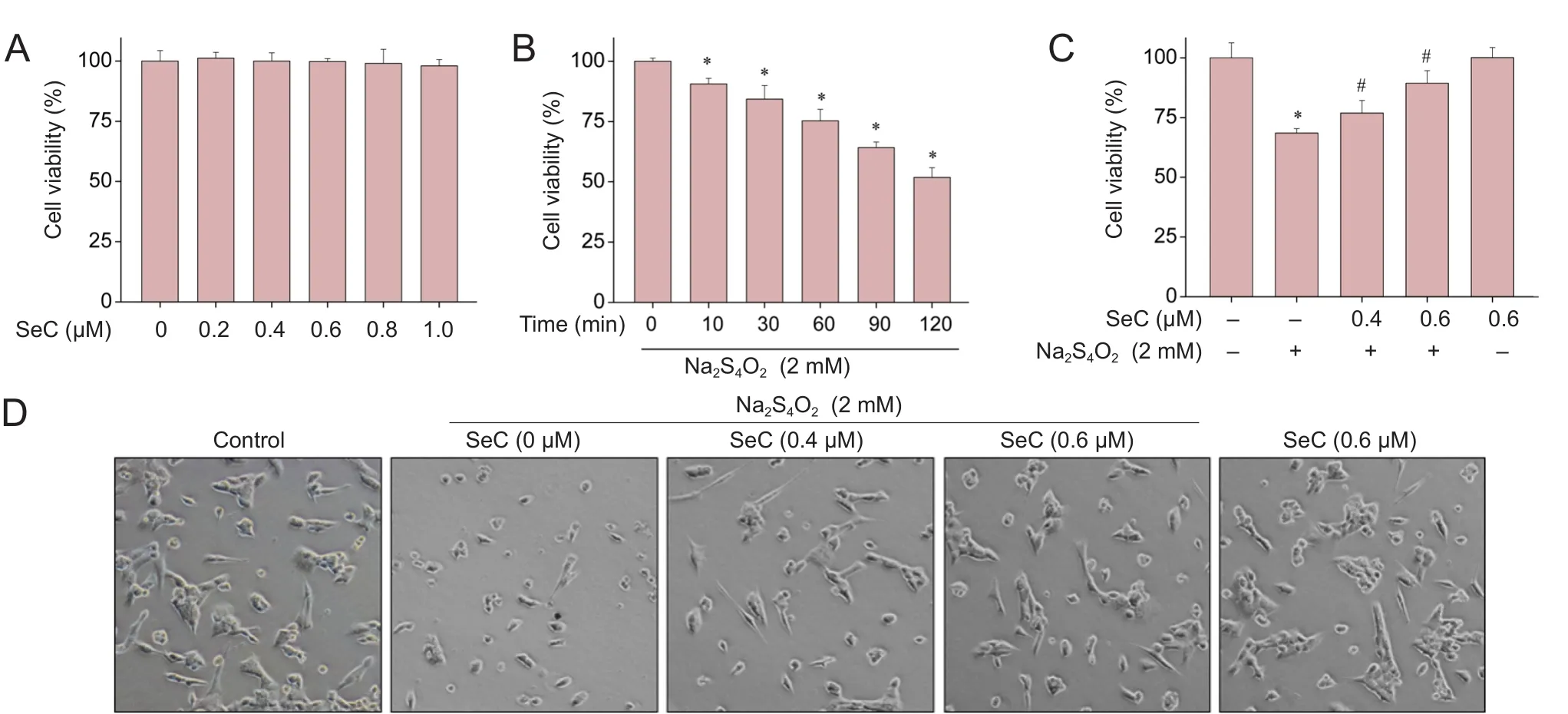
Figure 1 Selenocysteine (SeC) inhibits oxygen glucose deprivation (OGD)-induced cytotoxicity in HT22 cells.

Figure 2 Selenocysteine (SeC)suppresses oxygen glucose deprivation(OGD)-induced apoptosis in HT22 cells.
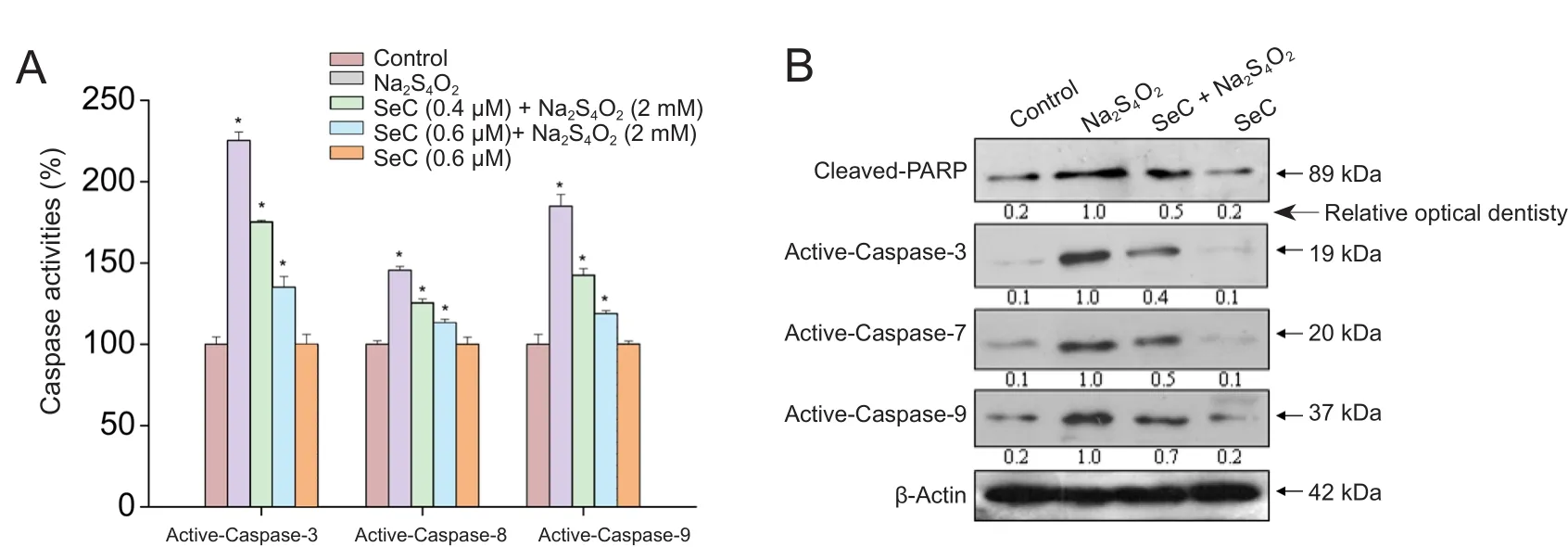
Figure 3 Effect of selenocysteine (SeC) on mitochondrial-mediated apoptosis in HT22 cells.
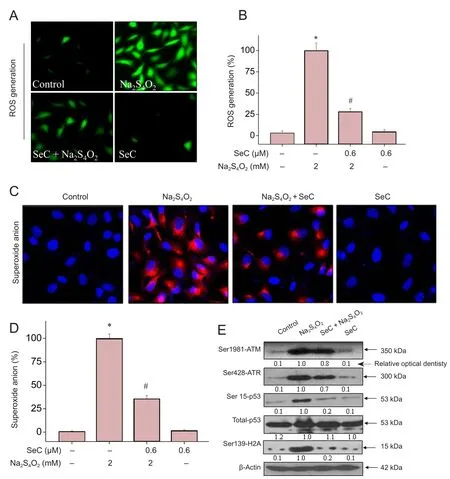
Figure 4 Selenocysteine (SeC)attenuates oxygen glucose deprivation (OGD)-induced oxidative damage by inhibiting reactive oxygen species (ROS) generation in HT22 cells.
SeC attenuates OGD-induced oxidative damage in HT22 cells
Oxidative damage acts as an important cell death mech‐anism in response to OGD treatment. Therefore, we also evaluated intracellular oxidative status in the current study.As shown in Figure 4A, B, cells cultured under OGD con‐ditions (glucose‐free medium + 2 mM Na2S4O2) showed a marked increase in ROS generation. Furthermore, ac‐cumulation of the superoxide anion was also detected in OGD‐treated cells (Figure 4C, D).
Furthermore, we found that ROS overproduction subse‐quently triggered DNA damage. As shown in Figure 4D, cells cultured under OGD conditions exhibited substantial DNA damage, as shown by the increased phosphorylation level of Ser1981‐ATM, Ser428‐ATR, Ser139‐H2A and Ser15‐p53.However, SeC pre‐treatment significantly attenuated OGD‐induced DNA damage by inhibiting ROS and superox‐ide anion generation (Figure 4E). Taken together, these re‐sults indicate that SeC has the potential to attenuate OGD‐in‐duced oxidative damage by inhibiting ROS generation.
Discussion
In the present study, HT22 mouse hippocampal neurons were cultured and employed to investigate the neuroprotec‐tive effect of SeC against OGD‐induced neurotoxicity. Com‐pared with 95% N2and 5% CO2, the use of Na2S4O2in a glu‐cose‐free medium is an effective way of quickly establishing OGD conditions, and is therefore commonly used in basic re‐search. Selenium, an essential trace element that is bene ficial in the chemoprevention and chemotherapy of cerebral isch‐emic stroke, is incorporated into. SeC, a selenium‐containing amino acid that also exhibits novel biological potential (Zhang et al., 2008; Chen and Wong, 2009). However, to date, it had not been investigated whether SeC antagonizes OGD‐in‐duced neurotoxicity in HT22 mouse hippocampal neurons.In the current study, thein vitroprotective potential and mechanism of selenocystine against oxygen glucose depriva‐tion (OGD) condition‐induced neurotoxicity were evaluated in HT22 mouse hippocampal neurons. Our findings con firm that SeC has the potential to ameliorate OGD‐induced neuro‐nal apoptosis, which has been shown to be the main cause of cell death in response to cerebral ischemia (Charriaut‐Mar‐langue et al., 1998; Snider et al., 1999; Mehta et al., 2012;Wu et al., 2013; Wang et al., 2016c). Apoptosis is an ordered cellular death process which can alter intracellular homeo‐stasis, and is controlled by serial gene and protein expression(Wu et al., 2013; Yuan et al., 2013; Fu et al., 2015, 2016). The extrinsic/death receptor‐mediated pathway and the intrinsic/mitochondrial‐mediated pathway are the two key pathways responsible for regulating cell apoptosis (Yagami et al., 2014;Fu et al., 2015; Li et al., 2015; Zhu et al., 2016). In this study,OGD‐induced activation of caspase‐8 and ‐9 was detected,which was effectively inhibited by SeC pre‐treatment. Activa‐tion of caspase‐9 was greater than that of casepase‐8, suggest‐ing that OGD‐induced apoptosis was mediated by the intrin‐sic apoptosis pathway. Moreover, activated caspase‐3 evoked a cell apoptosis cascade by inducing PARP cleavage. These mechanisms indicate that SeC interferes with OGD‐induced apoptosis mainly by inhibiting the mitochondrial‐mediated apoptotic pathway.
Superoxide anion, hydroxyl radical, and hydrogen per‐oxide are all involved in cerebral ischemia and contribute to ischemic neuronal damage (Floyd and Carney, 1992).Human brain tissue is extremely sensitive to oxidative damage due to its high oxygen consumption, high level of polyunsaturated fatty acids (Islam et al., 2002), and inability of nerve cells to regenerate (Floyd and Carney, 1992). There‐fore, suppression of ROS generation is crucial to preventing ischemic injury. P53, a transcription factor, acts as a tumor suppressor protein to regulate apoptosis, cell proliferation,cell cycle checkpoints, and DNA damage repair by regulat‐ing multiple signal transduction pathways (Aminzadeh et al., 2014, Renaud et al., 2014; Fan et al., 2017c). Activation of P53 through phosphorylation can induce apoptosis by activating downstream proteins, such as Bax/Bcl2, Fas/Apol,and IGF‐BP3 (Jarolim et al., 2017; Wang et al., 2017b; Beyfuss and Hood, 2018; Ma et al., 2018). Our findings show that over‐accumulation of ROS and superoxide anion activates DNA damage markers, including Ser139‐H2A and Ser15‐p53 in HT22 cells after treatment with Na2S4O2. However, SeC treatment reduced the production of free radicals and rescued HT22 cells from Na2S4O2‐induced DNA damage.
In conclusion, our findings provide evidence that SeC inhibits OGD‐induced oxidative damage and apoptosisin vitro. To our knowledge, this study is the first to fully inves‐tigate the molecular mechanisms underlying the action of SeC against OGD‐induced neurotoxicity. However, given that the present study focused solely onin vitroexperiments,future investigations of thein vivoeffects in animal models are warranted to clarify the mechanisms by which SeC pro‐tects against cerebral ischemic damage.
Acknowledgments:Appreciation of the experimental platform of School of Basic Medicine, Taishan Medical University, China.
Author contributions:XJW and XYF conceived the whole study.MHW, XTF, WC, YJH, SYB and DCT performed the experiments.MHW analyzed the results. XJW and XYF wrote the paper. All authors read and approved the final version of this paper for publication.
Conflicts of interest:The authors declare that there is no con flict of interest for all the authors.
Financial support:The study was supported by Sci-Tech Development Project of Taian in Shandong, No. 2016NS1058&2015NS2081; and the Sci-Tech Development Project of Linyi in Shandong, No. 201515006. The funders did not participant in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, and in the decision to submit the paper for publication.
Institutional review board statement:All animal experiments and procedures were conducted in accordance with the Ethics Committee of Taishan Medical University (approval No. 2016036).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Nobuyuki Ishibashi, Children’s National HealthSystem, USA; N. Scott Litofsky, University of Missouri-Columbia School of Medicine, USA.
Additional file:Open peer review reports 1 and 2.
- 中国神经再生研究(英文版)的其它文章
- In Memoriam: Ray Grill (1966–2018)
- Reorganization of injured anterior cingulums in a hemorrhagic stroke patient
- A novel chronic nerve compression model in the rat
- Analgesic effect of AG490, a Janus kinase inhibitor, on oxaliplatin-induced acute neuropathic pain
- Three-dimensional visualization of the functional fascicular groups of a long-segment peripheral nerve
- Novel conductive polypyrrole/silk fibroin scaffold for neural tissue repair