
Neuroprotective effects of genistein on SH-SY5Y cells overexpressing A53T mutant α-synuclein
2018-07-23 02:58HuanChengWuQunLiangHuShiJunZhangYanMinWangZhanKuiJinLingFuLvSaiZhangZhenLinLiuHongLianWuOuMeiCheng
中国神经再生研究(英文版) 2018年8期
Huan-Cheng Wu , Qun-Liang Hu , Shi-Jun Zhang Yan-Min Wang Zhan-Kui Jin Ling-Fu Lv Sai Zhang , Zhen-Lin Liu ,Hong-Lian Wu , Ou-Mei Cheng
Abstract Genistein, a potent antioxidant compound, protects dopaminergic neurons in a mouse model of Parkinson’s disease. However, the mecha‐nism underlying this action remains unknown. This study investigated human SH‐SY5Y cells overexpressing the A53T mutant of α‐synuclein.Four groups of cells were assayed: a control group (without any treatment), a genistein group (incubated with 20 μM genistein), a rote‐none group (treated with 50 μM rotenone), and a rotenone + genistein group (incubated with 20 μM genistein and then treated with 50 μM rotenone). A lactate dehydrogenase release test con firmed the protective effect of genistein, and genistein remarkably reversed mitochondrial oxidative injury caused by rotenone. Western blot assays showed that BCL‐2 and Beclin 1 levels were markedly higher in the genistein group than in the rotenone group. Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling revealed that genistein inhibited rotenone‐induced apoptosis in SH‐SY5Y cells. Compared with the control group, the expression of NFE2L2 and HMOX1 was signi ficantly increased in the genistein + rotenone group. However, after treatment with estrogen receptor and NFE2L2 channel blockers (ICI‐182780 and ML385, respectively), genistein could not elevate NFE2L2 and HMOX1 expression. ICI‐182780 ef‐fectively prevented genistein‐mediated phosphorylation of NFE2L2 and remarkably suppressed phosphorylation of AKT, a protein downstream of the estrogen receptor. These findings con firm that genistein has neuroprotective effects in a cell model of Parkinson’s dis‐ease. Genistein can reduce oxidative stress damage and cell apoptosis by activating estrogen receptors and NFE2L2 channels.
Key Words: nerve regeneration; genistein; neuroprotection; SH-SY5Y cells; Parkinson’s disease; rotenone; estrogen receptor; NFE2L2; A53T;α-synuclein; oxidative stress; neurodegeneration; neural regeneration
Introduction
Parkinson’s disease (PD) is a neurodegenerative disease characterized by severe motor deficits, including bradyki‐nesia, resting tremor, postural instability and rigidity. The pathophysiological changes responsible for these motor de fi‐cits are associated with the selective death of dopaminergic neurons located in the substantia nigra pars compacta and the subsequent decrease of striatal dopamine content. Al‐though L‐dopa or monoamine oxidase B inhibitors, such as rasagiline, provide symptomatic relief, no available therapy can delay or halt the neurodegenerative process of PD (Chai et al., 2013; Lim et al., 2013; Buard et al., 2016). Therefore, an urgent clinical need exists for effective PD drugs and ther‐apies. Effective therapies for PD may be achieved by target‐and mechanism‐based drug development (Trippier et al.,2013; Khanam et al., 2016). Previous studies have consistently indicated that oxidative stress and mitochondrial dysfunction are common mechanisms that lead to the demise of dopa‐minergic neurons in both familial and sporadic PD (Hwang,2013; Yan et al., 2013; Kim et al., 2016). Rotenone is a toxin that selectively kills dopaminergic neurons (Lehmensiek et al., 2006; Ximenes et al., 2015; Jeitner et al., 2016). Once inside the neuron, rotenone produces hydrogen peroxide,superoxide, and hydroxyl radicals. This process causes lipid peroxidation and DNA oxidation and finally results in mito‐chondrial oxidative stress, mitochondrial dysfunction, and apoptosis (Xu et al., 2012; Hwang, 2013; Nataraj et al., 2017).Neuroprotective therapies for PD are presumed to suppress oxidative stress and reverse mitochondrial dysfunction. We mimicked the pathogenesis of PDin vitroby treating SH‐SY5Y cells overexpressing A53T mutant α‐synuclein with rotenone (Alberio et al., 2010; Ouzounoglou et al., 2014). This model of PD is widely used for studies of neuronal survival and apoptosis (Liu et al., 2015) and these cells can succumb to oxidative stress, mitochondrial dysfunction, and apoptosis(Li et al., 2014; Guardia‐Laguarta et al., 2015; Paillusson et al.,2017). This model can also be used to identify neuroprotec‐tive agents and probe their underlying mechanisms.
Genistein (GS), the main active ingredient of Genista tinctoria, has neuroprotective properties against PD (Luo et al., 2011). Our preliminary experiments found that GS can protect neurons against various toxic stimuli; therefore, in the present study, multiple approaches were conducted to investigate the neuroprotective actions of GS and the under‐lying mechanisms.
Heme Oxygenase 1 (HMOX1) belongs to the phage 2 detoxifying enzymes, and together with glutathione perox‐idase, plays a central role in neuronal protection (Barlow et al., 2005; Büeler 2009). They can be up‐regulated in neuronsviathe activation of nuclear factor‐erythroid 2‐related factor 2 (NFE2L2) (Du et al., 2013; Kulasekaran et al., 2015; Kwon et al., 2015). Targeting NFE2L2 signaling may provide neu‐roprotection advantages. Furthermore, NFE2L2 activation is regulated by multiple signaling pathways, such as estrogen receptor (Zuo et al., 2013; Lopert et al., 2016; Li et al., 2017)and phosphoinositide 3‐kinase/AKT pathways (Zoubeidi et al., 2010; Bowser et al., 2011; Xu et al., 2011; Wang et al., 2011; Sun et al., 2012; Park et al., 2013; Li et al., 2015).Whether these mechanisms are responsible for GS‐mediated neuroprotection remains to be seen.
To investigate the mechanism of GS neuroprotection as a promising therapeutic target for PD, we performedin vitroexperiments.
Materials and Methods
Cell culture
SH‐SY5Y cells overexpressing A53T mutant α‐synuclein (Cell Resource Center of the Institute of Basic Medical Sciences,Peking Union Medical College/Chinese Academy of Medical Sciences, Beijing, China) were maintained in a 1:1 mixture of F12 nutrient medium (Sigma, St. Louis, MO, USA) and Dulbecco’s modified Eagle’s medium (DMEM, Gibco Life Technologies, Grand Island, NY, USA) supplemented with 10% (v/v) heat‐inactivated fetal bovine serum (Gibco Life Technologies), 2 mM L‐glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin (Sigma). The cells were grown in DMEM (Gibco Life Technologies) containing 10% heat‐inac‐tivated horse serum (Sigma), 5% heat‐inactivated fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin. The cells were maintained at 37°C in a humidi fied atmosphere of 95% air and 5% CO2. Cells below passage 10 and in the expo‐nential growth phase were used in all experiments.
Drug preparation
GS (Shanghai Winherb Medical S&T Development, Shang‐hai China) was stored at 4°C as a stock solution (100 mM) in dimethyl sulfoxide (Sigma). Rotenone (Sigma) was dissolved in sterile distilled water containing 0.1% ascorbic acid as a stock solution (1 M). GS and rotenone stock solutions were diluted in DMEM/F‐12 immediately before use.
Four cell groups were prepared. The control group was without any treatment. Cells in the rotenone group were treated with 50 μM rotenone for 24 hours. Cells in the GS group were incubated with 20 μM GS for 24 hours. Cells in the rotenone + GS group were incubated with 20 μM GS for 24 hours and then treated with 50 μM rotenone for 24 hours.
Analysis of cell viability and morphological changes
Cell viability was determined using cell a counting kit‐8 kit(Dojindo Laboratories, Kyushu, Japan). A53T mutant α‐sy‐nuclein‐overexpressing SH‐SY5Y cells were cultured in 96‐well plates at a density of 1 × 104cells/well and grown for 24 hours. The cells were treated with rotenone for 24 hours or pre‐incubated with GS for 24 hours and then incubated with or without rotenone for 24 hours. Cells incubated in DMEM/F12 containing an equivalent concentration of di‐methyl sulfoxide (the highest concentration was less than 0.1%) were used as control cells. Thereafter, 10 μL of cell counting kit‐8 solution was added to each well. The absor‐bance was detected at 450 nm on a microplate reader after 1 hour (SpectraFluor, Tecan, Sunrise, Austria). Cell viability was expressed as a percentage of the control. Morphologi‐cal changes were observed under a light microscope (Aote,Chongqing, China).
Determination of lactate dehydrogenase (LDH) activity
LDH was measured using a lactate dehydrogenase assay kit according to the manufacturer’s protocol (Nanjing Ji‐ancheng Bioengineering Institute, Nanjing, China). SH‐SY5Y cells at 1 × 105cells/well were cultured in 6‐well plates.The groups were prepared as above. The culture medium was collected and the level of extracellular LDH measured.The cells were harvested to measure the level of intracellular LDH activity.
Detection of cell DNA fragmentation with terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)
DNA fragmentation in apoptotic SH‐SY5Y cells was detect‐ed by the TUNEL assay by using the ApopTag Fluoresce In Situ Apoptosis Detection Kit (Millipore, Billerica, MA,USA). SH‐SY5Y cells were cultured on cover slips. The groups and the interventions were as described above. After treatment, cells were washed twice with PBS and fixed in 4%neutral‐buffered formalin solution for 30 minutes. The cells were then rinsed with PBS and incubated with a methanol solution containing 0.3% H2O2for 30 minutes. Thereafter,the cells were incubated in a permeabilizing solution (0.1%sodium citrate and 0.1% Triton X‐50) for 10 minutes. After rinsing in equilibration buffer, the cells were incubated with a working‐strength TdT enzyme in a humidi fied chamber at 37°C for 1 hour. The cells were then rinsed in the stop/wash buffer and incubated with the working‐strength anti‐digoxi‐genin conjugate at room temperature for 30 minutes. After washing in PBS, the cells were counterstained with diamidi‐no‐2‐phenylindole (DAPI) and viewed under a fluorescence microscope (Leica, Solms, Germany). For quantitative anal‐ysis, positively stained cells from five random visual fields(100 μm × 100 μm) were counted, and the result shown as the ratio of positively stained cells to total cells.
Determination of oxidation in mitochondria
Oxidation in mitochondria was determined after rotenone treatment for 24 hours using a glutathione kit, a malondialde‐hyde (MDA) kit, an adenosine triphosphate (ATP) kit, and a Na+‐K+‐ATPase kit (all from Nanjing Jiancheng Bioengineer‐ing Institute) according to the manufacturer’s instructions.Results were visualized on photo‐sensitive film (Amersham Biosciences Ltd., Chalfont St. Giles, UK) using enhanced che‐miluminescence reagents (Pierce, Rockford, IL, USA).
Estrogen receptor and NFE2L2 inhibition
Estrogen receptor inhibitor (ICI‐182780) and NFE2L2 in‐hibitor (ML385) were purchased from Santa Cruz Biotech‐nology (Santa Cruz, CA, USA). SH‐SY5Y cells were cultured in 6‐well plates. To investigate the estrogen receptor and the NFE2L2 pathway, the cells were divided into six groups: a control group, a rotenone group, a GS group and a rotenone+ GS group were treated as above; the M and ICI groups were treated with ICI182780 (100 nM) and ML385 (50 nM),respectively, for 24 hours. The M + GS and ICI + GS groups were pre‐treated with 20 μM GS and then treated with ICI182780 (100 nM) and ML385 (50 nM), respectively, for 24 hours.
Western blot assays
Protein levels were determined after rotenone treatment for 24 hours by western blot assays. Briefly, equal amounts of protein were separated by electrophoresis in 10% sodium dodecyl sulfate polyacrylamide gels and transferred onto ni‐trocellulose membranes. The membranes were incubated with 5% (w/v) non‐fat milk in Tris‐buffered saline containing 0.1%(v/v) Tween 20 for 2 hours to block nonspeci fic binding. The membranes were then incubated overnight at 4°C with pri‐mary antibodies. The primary antibodies used were as follows:rabbit polyclonal anti‐estrogen receptor (1:2000); rabbit poly‐clonal anti‐NFE2L2 (1:2000); rabbit polyclonal anti‐Lamin B1 (1:1000); goat polyclonal anti‐P‐BAD (1:1000); rabbit polyclonal anti‐BAD (1:2000); mouse monoclonal anti‐BCL‐2(anti‐apoptotic protein) (1:500); rabbit polyclonal anti‐BAX(apoptotic protein) (1:2000); mouse monoclonal anti‐Beclin 1(anti‐apoptotic protein) (1:500); goat polyclonal anti‐cleaved caspase 3 (apoptotic protein) (1:1000); mouse monoclonal anti‐HMOX1 (1:2000); rabbit polyclonal anti‐AKT (1:2000),rabbit polyclonal anti‐phospho‐AKT (1:2000); and mouse monoclonal anti‐GAPDH (1:1000) (all from Santa Cruz Biotechnology). After washing with Tris‐buffered saline and Tween 20, the membranes were incubated for 1 hour at room temperature with peroxidase‐conjugated secondary antibod‐ies. The membranes were washed again with Tris‐buffered saline and Tween 20, and the immunolabeling developed using an enhanced chemiluminescence reagent (Sigma). The protein levels were quanti fied by densitometry using Image J software (https://imagej.nih.gov/ij/).
Statistical analysis
The data are expressed as the mean ± SD of three indepen‐dent experiments. One‐way analysis of variance followed by Student-Newman-Keulspost hoctest was performed for multiple group comparisons and two‐group comparisons using GraphPad Prism 6.0 software (GraphPad Software Inc., CA, USA). A value ofP< 0.05 was considered statisti‐cally signi ficant.
Results
GS inhibited rotenone-induced cell death in SH-SY5Y cells overexpressing A53T mutant α-synuclein
The potential protective effects of GS were investigated in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein exposed to rotenone. First, the effect of rotenone was in‐vestigated in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein. The treatment of SH‐SY5Y cells with different concentrations (25, 50, 100, and 200 μM) of rotenone for 4,8, 16, 24, and 32 hours decreased cell viability in a concen‐tration‐ and time‐dependent way (Figure 1A). Cell viability was reduced to approximately 50% of the control when the cells were exposed to 50 μM of rotenone for 24 hours. Thus,the concentration of 50 μM and period of 24 hours were used for further investigations.
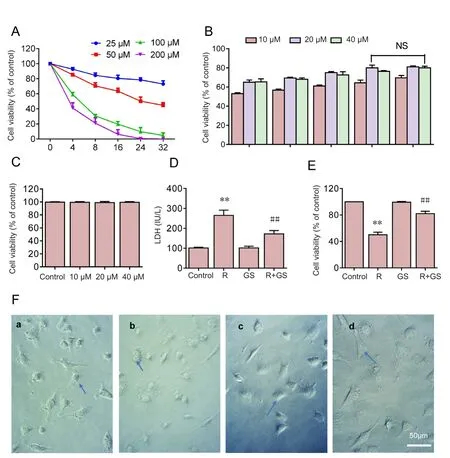
Figure 1 GS inhibits rotenoneinduced cell death in SH-SY5Y cells overexpressing A53T mutant α-synuclein.
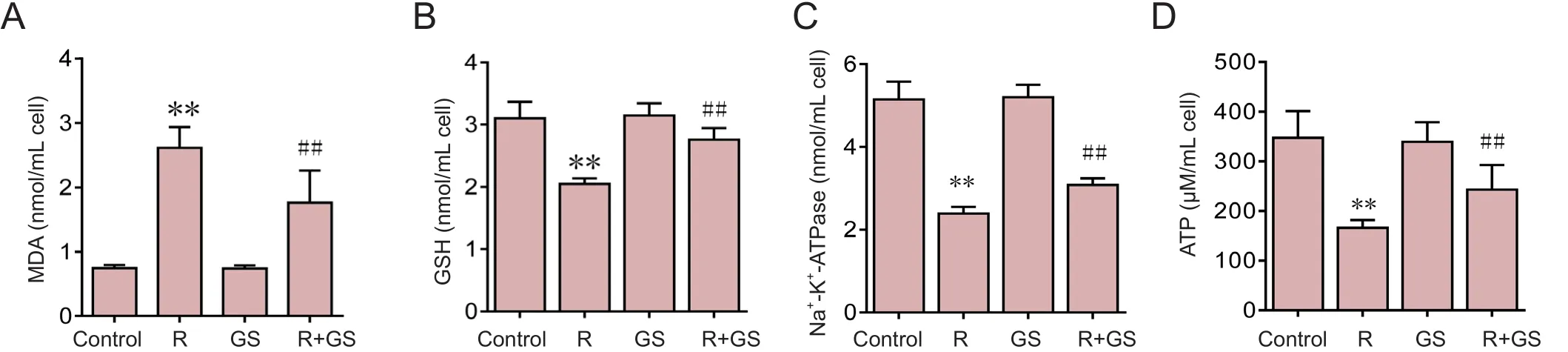
Figure 2 GS inhibits rotenone-induced mitochondrial oxidative stress.
Second, the potential protective effect of GS on rote‐none‐induced cell death in SH‐SY5Y cells was explored. The pre‐incubation of the cells with increasing concentrations(10, 20, and 40 μM) of GS for different time periods (4, 8, 16,and 24 hours) reversed the decreased cell viability caused by rotenone treatment. However, GS pre‐incubation of the cells with 40 μM GS for 32 hours had no increased effect of pro‐tection compared with 20 μM GS for 24 hours, and the cell viability remained at 4/5 of control viability. This result sug‐gested that the maximum protective in fluence was obtained with 20 μM GS for 24 hours. Therefore, the concentration of 20 μM and time period of 24 hours were selected for further investigations (Figure 1B).
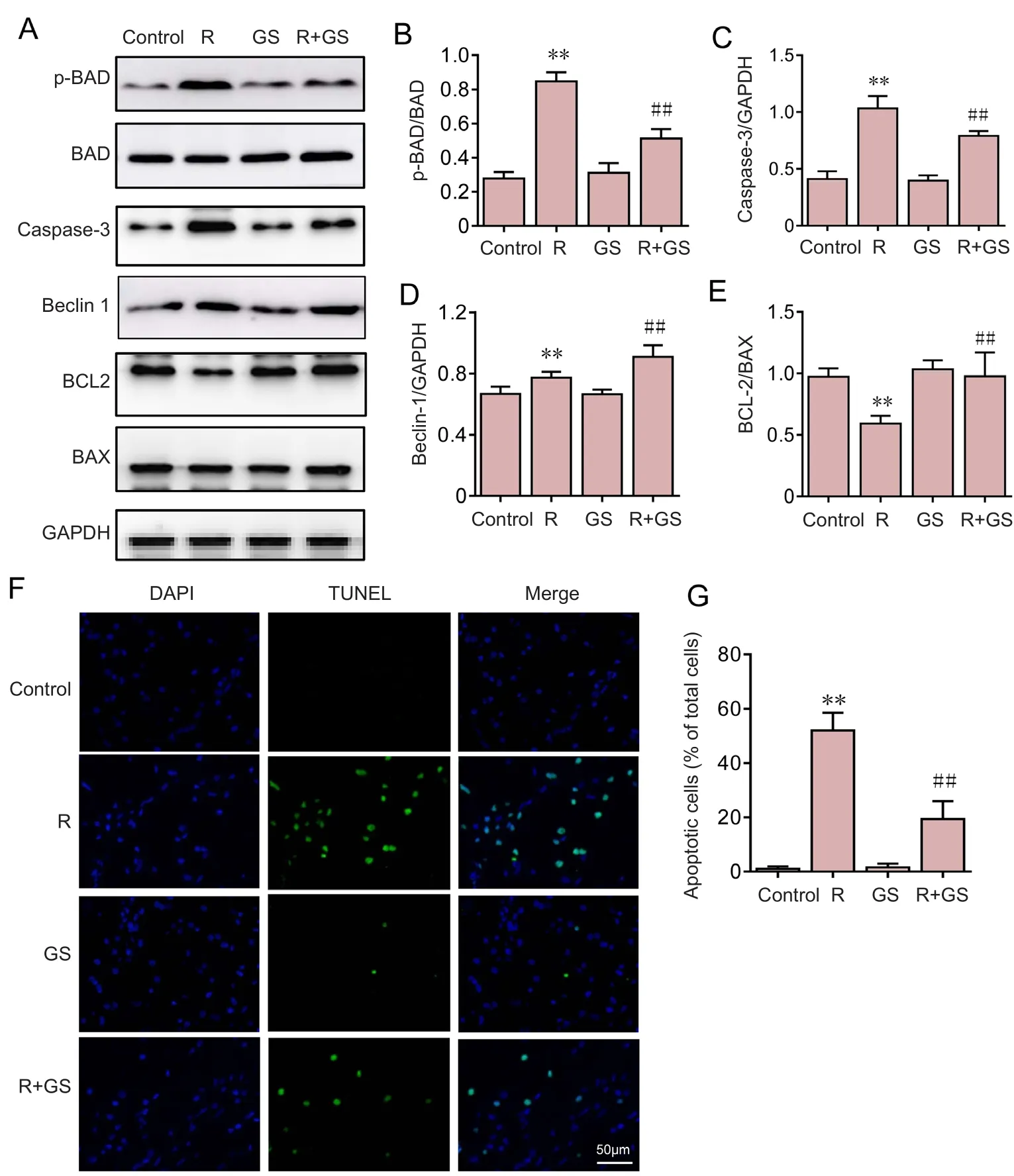
Figure 3 GS inhibition of rotenone-induced apoptosis in SH-SY5Y cells.

Figure 4 GS activated NFE2L2 pathways in SH-SY5Y cells.
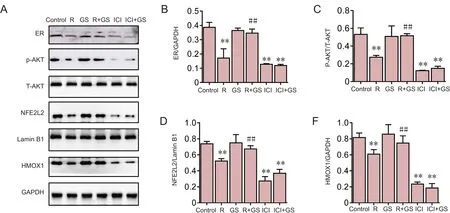
Figure 5 Dependence of GS-mediated NFE2L2 activation on ER pathways.
Third, the effect of GS on SH‐SY5Y cells overexpressing A53T mutant α‐synuclein was investigated. No significant difference in cell viability was found when the cells were incu‐bated for 24 hours with different concentrations (10, 20, and 40 μM) of GS compared with the control cells (P= 0.928). This finding indicated that GS had no toxic effect on SH‐SY5Y cells overexpressing A53T mutant α‐synuclein (Figure 1C).
The protective effect of GS was con firmed using an LDH release assay. Pre‐incubation of SH‐SY5Y cells with 20 μM GS for 24 hours decreased LDH release in cells treated with rotenone compared with the rotenone group (P= 0.0067).By contrast, GS pre‐treatment without rotenone did not in‐fluence LDH release (Figure 1D).
Similar to the LDH release results, pre‐incubation with GS protected cells as assessed using the cell counting kit‐8 test.Cell viability was increased with GS (20 μM) pre‐incubation for 24 hours compared with the rotenone group (P= 0.0005).This finding indicated that GS pretreatment exerted a pro‐tective effect on rotenone treated cells.
There was no remarkable difference in cell morphology between GS and control groups under light microscopy ex‐amination (Figure 1Fa, c). The rotenone group showed obvi‐ous partial cell lysis. The number of cells decreased, the cells were irregular in shape, and the number of cell synapses de‐creased (Figure 1Fb). However, GS pretreatment remarkably inhibited cell damage induced by rotenone (Figure 1Fd).
GS inhibited rotenone-induced mitochondrial oxidative stress
Compared with the control group, MDA content, glutathi‐one content, ATP content, and Na+‐K+‐ATPase activity were reduced in the rotenone group (P< 0.05) (Figure 2). With rotenone treatment, antioxidant levels decreased; oxidative damage gradually occurred; mitochondrial capacity was blocked; and ATP production gradually diminished, result‐ing in a gradual reduction of enzyme activity (Figure 2).However, GS pre‐treatment increased glutathione content,ATP content, and Na+‐K+‐ATPase activity and decreased MDA content, compared with the rotenone group (P< 0.05)(Figure 2). GS signi ficantly reversed the mitochondrial oxi‐dative damage induced by rotenone.
GS inhibited rotenone-induced apoptosis in SH-SY5Y cells
To investigate whether GS inhibits rotenone‐induced apop‐tosis, p‐BAD, BAD, caspase 3, Beclin 1, BCL-2 and BAX were analyzed using western blot assays (Figure 3A). Com‐pared with the control group, P‐BAD, caspase 3 and Beclin 1 expression were signi ficantly increased in the rotenone group(P< 0.01) (Figure 3B–D), but BCL‐2 expression was signi fi‐cantly decreased in the rotenone group (P< 0.01; Figure 3E),indicating that apoptosis was induced by rotenone. However,the BAD and caspase 3 levels were decreased in the GS group compared with the rotenone group (P< 0.01) (Figure 3B–D),but BCL‐2 and Beclin 1 levels were signi ficantly higher in the GS group than in the rotenone group (P< 0.05; Figure 3D, E),which demonstrates that GS has an inhibitory effect on rote‐none‐induced apoptosis in SH‐SY5Y cells.
DNA fragmentation is a typical marker of apoptosis.Therefore, nuclear fragmentation in apoptotic cells was detected to investigate the effects of GS on rotenone‐in‐duced apoptosis. DNA fragmentation and the number of TUNEL‐positive cells were dramatically augmented in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein exposed to rotenone compared with the control group. By contrast, pretreatment with GS effectively reversed these changes induced by rotenone, but GS alone had no effect on DNA fragmentation (Figure 3F). This result indicated that the apoptosis rate was signi ficantly increased when the cells were challenged by rotenone (Figure 3G) and that these changes were markedly reversed by GS pre‐incubation. GS treatment without rotenone had no effect on the apoptosis rate. These results indicated that GS was capable of rescuing SH‐SY5Y cells overexpressing A53T mutant α‐synuclein from rotenone‐induced apoptotic death.
GS activated NFE2L2 pathways in SH-SY5Y cells
Given that NFE2L2 activation has bene ficial effects on cell survival, we examined the effect of GS on NFE2L2 and on activation of its downstream protein HMOX1 in SH‐SY5Y cells by western blot assays (Figure 4A). SH‐SY5Y cells treated with GS exhibited enhanced levels of nuclear NFE2L2 accumulation and HMOX1 activity (Figure 4B, C).However, compared with the control group, the activation of NFE2L2 and HMOX1 was significantly inhibited in the rotenone group (P< 0.01). Pretreatment with GS reduced the inhibitory effect of rotenone on NFE2L2 and HMOX1 (P< 0.01). However, after blocking NFE2L2 with the inhibitor ML385, GS did not promote activation of the downstream protein HMOX1 (Figure 4C).
Dependence of GS-mediated NFE2L2 activation on estrogen receptor pathways
To delineate the pathway involved in GS‐mediated NFE2L2 activation, SH‐SY5Y cells overexpressing A53T mutant α‐synuclein were pretreated with estrogen receptor inhib‐itor, ICI‐182780 (20 μM). The activation of NFE2L2 and HMOX1 was assessed by western blot detection of the estro‐gen receptor and phosphorylation of its downstream pro‐tein, AKT. ICI‐182780 effectively prevented the upregulated level of NFE2L2 mediated by GS (Figure 5A–E). Inhibition of the estrogen receptor signi ficantly suppressed the phos‐phorylation of AKT (P< 0.01; Figure 5B). The GS‐mediated enhancement of NFE2L2 and HMOX1 activity was also abolished by treating SH‐SY5Y cells with ICI‐182780 (P<0.01) (Figure 5D, E). GS activates NFE2L2 signalingviaes‐trogen receptor activation in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein.
Discussion
PD is a neurodegenerative disease that causes the selective death of dopaminergic neurons in the substantia nigra.Although the etiology and pathogenesis of PD are not com‐pletely elucidated, accumulating evidence indicates that ro‐tenone, a hydroxylated dopamine metabolite, contributes to neuronal cell death in PD (Andrew et al., 1993; Chambers et al., 2013; Leal et al., 2016; Peng and Zhao, 2016).
Rotenone can produce superoxide, hydroxyl radicals, and hydrogen peroxide, which can cause lipid peroxidation and DNA oxidation resulting in mitochondrial oxidative stress,mitochondrial dysfunction, and apoptosis (Hwang, 2013;Nataraj et al., 2017; Rocha et al., 2017). Rotenone is widely accepted as a toxin for the induction ofin vivoandin vitroPD models (Sriraksa et al., 2012; Chambers et al., 2013;Tovilovic et al., 2013). This study investigated the neuropro‐tective effect of GS and the underlying mechanisms using anin vitromodel of rotenone‐induced cell death in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein. This model bypasses any possible physiological feedback interactions of anin vivomodel.
Rotenone caused oxidative stress in SH‐SY5Y cells overex‐pressing A53T mutant α‐synuclein, which is consistent with previous studies (Cunha et al., 2013; Pasban‐Aliabadi et al.,2017). Oxidative stress proceeded after the cells’ anti‐oxida‐tive ability was saturated by the production of reactive oxy‐gen species. In this study, an increase in MDA levels and a striking decrease in the activities of HMOX1 were observed in rotenone‐treated cells. However, these rotenone effects were suppressed by GS pre‐incubation.
HMOX1 has a central function in neuroprotection against oxidative stress. HMOX1 is a rate‐limiting enzyme that pro‐motes the oxidative catabolism of heme and produces biliver‐din, carbon monoxide, and ferrous iron. HMOX1 expression is controlled by an important transcription factor, NFE2L2.In the basal state, NFE2L2 is sequestered in the cytoplasm with NFE2L2‐Kelch‐like ECH‐associated protein 1 (Keap1).When activated, NFE2L2 detaches from the NFE2L2—Keap1 complex and translocates to the nucleus. NFE2L2 then com‐bines with the antioxidant responsive element to induce the expression of phage 2 detoxifying enzymes (Kim et al., 2008;Kang et al., 2015; Jeong et al., 2016). We found that the nucle‐ar translocation of NFE2L2 in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein increased in response to GS stim‐ulation in a time‐dependent manner. The actions of HMOX1 also increased in a time‐dependent manner in response to GS incubation. To determine whether elevation of HMOX1 activ‐ity is dependent on NFE2L2 activation, we treated cells with the NFE2L2 inhibitor, ML385. The GS‐mediated up‐regula‐tion of HMOX1 activity was effectively inhibited by ML385.The neuroprotective effect of GS was also partly suppressed by ML385. Moreover, the level of NFE2L2 was down‐regulat‐ed by rotenone, as were the activities of HMOX1. However,GS pre‐incubation effectively inhibited these changes induced by rotenone. These results indicate that the neuroprotective effect of GS results partly from NFE2L2‐dependent enhance‐ment of HMOX1.
Our results are consistent with those of a previous study in which rotenone resulted in mitochondrial dysfunction and apoptosis in SH‐SY5Y cells overexpressing A53T mutant α‐synuclein (Nataraj et al., 2017). This was manifested by mitochondrial oxidative stress, caspase‐3 activation, DNA fragmentation, and an increased rate of apoptosis. However,GS was effective in protecting SH‐SY5Y cells overexpressing A53T mutant α‐synuclein against rotenone‐induced apopto‐sis. This is the first study to report the neuroprotective effect of GS against rotenone‐induced apoptosis. The deregulation of BCL‐2 is responsible for the mitochondrial dysfunction induced by rotenone. In the presence of apoptotic factors,the pro‐apoptotic protein BAD interacts with the anti‐apop‐totic protein BCL‐xl, thereby releasing the pro‐apoptotic protein BAX from the BCL‐xl—BAX complex. BAX can disrupt the mitochondrial membrane, causing the release of cytochrome c from the mitochondria. Consequently,caspase‐3 is activated and apoptosis is initiated (Yang et al.,1995; Maurya et al., 2016; Wang et al., 2017). However, BAD can be inhibited when it is phosphorylated at serine 112.Therefore, promoting the phosphorylation of BAD at serine 112 contributes to apoptosis inhibition (Bonni et al., 1999;Gu et al., 2004). We therefore investigated the effect of GS on BAD. GS time‐dependently increased BAD phosphory‐lation, thereby preventing rotenone‐induced apoptotic cell death. To our knowledge, this is the first study showing GS neuroprotection against rotenone‐induced apoptosis.
Given that GS activated NFE2L2 in a time‐dependent manner, the fact that GS‐mediated protection was achieved by pre‐incubation only rather than co‐treatment with rote‐none was not surprising and can be attributed to the time needed for the transcription and translation needed. How‐ever, the mechanism of NFE2L2 activation is still unclear.Previous studies showed that estrogen receptor pathways might be involved in NFE2L2 activation in various cell types(Zoubeidi et al., 2010; Bowser et al., 2011; Xu et al., 2011;Wang et al., 2011; Sun et al., 2012; Park et al., 2013; Li et al., 2015). To ascertain which pathway participated in the activation of NFE2L2 mediated by GS, we used the estrogen receptor inhibitor, ICI‐182780. We found that ICI‐182780 achieved a nearly complete inhibition of NFE2L2 activa‐tion. This indicated that the estrogen receptor was involved in GS‐mediated NFE2L2 activation. Moreover, GS‐medi‐ated neuroprotection was partly inhibited by ML385 and completely abolished by ICI‐182780. These results indicate that the immediate upstream activator of NFE2L2 was the estrogen receptor, and that these factors are all involved in GS‐mediated neuroprotection either directly or indirectly.
Although NFE2L2 activation was clearly activated by estro‐gen receptor signaling pathways, the occurrence of cross talk between NFE2L2 and estrogen receptor was not clearly eluci‐dated. Detailed experiments are needed to investigate the exact underlying mechanism. Furthermore, the protective effect of GS on different neuronal cell types remains to be determined.Taken together, we have demonstrated the neuroprotective ef‐fects of GSin vitro. GS is therefore a promising candidate drug for PD treatment that may provide an alternative adjunctive medication for the treatment of this neurodegenerative dis‐ease. GS exhibited neuroprotective effects against rotenone‐in‐duced apoptosis in SH‐SY5Y cells. The mechanism of this neuroprotection was associated with GS‐mediated NFE2L2 activationviaestrogen receptor pathways.
Acknowledgments:We are grateful for the financial and equipment supported by the Center for Neurology and Neurosurgery of Affiliated Hospital, Logistics University of Chinese People’s Armed Police Force, China.
Author contributions:HCW and HLW performed statistical analysis and wrote the paper. QLH, SJZ, and YMW were involved in the manuscript preparation. HCW and ZLL were responsible for the study design and data collection. ZKJ and LFL performed some of the experiments.SZ was responsible for the study design and obtained the funding. All authors approved the final version of the paper.
Conflicts of interest:None declared.
Financial support:This study was supported by a grant from the National Key Research and Development Plan of China, No.2016YFC1101500; the National Natural Science Foundation of China, No.11672332, 11102235, 8167050417; the Key Science and Technology Support Foundation of Tianjin City of China, No. 17YFZCSY00620; the Natural Science Foundation of Tianjin City of China, No. 15JCYBJC28600,17JCZDJC35400. The funding bodies played no role in the study design,in the collection, analysis and interpretation of data, in the writing of the paper, and in the decision to submit the paper for publication.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Jin Zhang, Faculty, Dalhousie University, Canada.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- In Memoriam: Ray Grill (1966–2018)
- Structural brain volume differences between cognitively intact ApoE4 carriers and non-carriers across the lifespan
- Roles of Eph/ephrin bidirectional signaling during injury and recovery of the central nervous system
- Neuroplasticity, limbic neuroblastosis and neuro-regenerative disorders
- A tissue-engineered rostral migratory stream for directed neuronal replacement
- Targeting the noradrenergic system for anti-in flammatory and neuroprotective effects:implications for Parkinson’s disease