
黑龙江省东部大豆疫霉群体遗传多样性时空动态
2018-07-16 11:21:42文景芝赵钰琦高新颖宫丽娟丁俊杰陈宇飞
东北农业大学学报 2018年7期
文景芝,张 斌,田 苗,2,揭 岩,赵钰琦,高新颖,宫丽娟,顾 鑫,丁俊杰,陈宇飞
(1.东北农业大学农学院,哈尔滨 150030;2.黑龙江省农垦科学院农畜产品综合利用研究所,黑龙江 佳木斯 154007;3.黑龙江省农业科学院佳木斯分院,黑龙江 佳木斯 154007)
由大豆疫霉(Phytophthora sojae Kaufmann&Gerdemann)侵染引起的大豆疫霉根腐病(Phytophthora root and stem rot,PRR)于1948年在美国首次发现[1],随后日本、巴西、加拿大和阿根廷等国相继出现[2-3]。苏彦纯等在我国东北地区分离到大豆疫霉[4]。近年来,PRR在黑龙江省普遍发生[5]。
简单重复序列(Simple Sequence Repeat,SSR)分子标记具有多态性及重复性好、操作简便、所需DNA量少和稳定快速等特点[6],广泛应用于植物病原菌遗传多样性研究。Cooke等在爱尔兰马铃薯晚疫病菌遗传多样性研究中发现,马铃薯晚疫病菌[Phytophthora infestans(Mont.)de Bary]最早可能由英国传入[7];Banniza等利用SSR标记法分析科特迪瓦黄瓜立枯病菌(Rhizoctonia solani Kühn)遗传多样性,认为同一地理来源黄瓜立枯病菌具有显著遗传多样性[8];党悦嘉等利用SSR分子标记分析黄瓜霜霉病菌(Pseudoperonospora cubensis(Berk.et Curt.)Rostov)遗传多样性,表明中国江苏省、广东省和湖北省黄瓜霜霉菌遗传分化较大,群体遗传多样性丰富,SSR遗传多样性与菌株地理来源存在相关性[9]。Mohammadi等对伊朗大豆疫霉遗传多样性作SSR分析时发现,即使同一省份大豆疫霉群体也存在较大变异[10];徐静静等利用18对SSR引物分析黑龙江省、福建省和美国大豆疫霉遗传多样性,发现黑龙江省和福建省大豆疫霉遗传距离最近,美国和福建省大豆疫霉遗传距离最远[11];范勇利用SSR标记分析福建省和黑龙江省大豆疫霉遗传多样性,结果表明黑龙江省群体遗传变异最小[12];王子迎等在黑龙江省和安徽省大豆疫霉遗传多样性分析中发现,安徽省群体与黑龙江省群体间遗传相似性较低,但来源不确定[13];黑龙江省与湖北省大豆疫霉群体遗传距离较大[14]。
病原菌遗传多样性变化规律对培育抗病品种至关重要,培育抗病品种是防治大豆疫霉根腐病最经济有效方法。目前,黑龙江大豆疫霉遗传多样性报道较多,但多为单一地块或某一年不同地区大豆疫霉遗传多样性分析,难以揭示遗传多样性规律。
本试验连续4年在大豆疫霉根腐病重病区黑龙江省东部试验田和生产田定点采集土壤样品并分离大豆疫霉,利用SSR分子标记方法分析大豆疫霉遗传多样性,以阐明黑龙江省东部大豆疫霉群体遗传多样性随时间和空间变化规律,为利用抗病品种防治大豆疫霉根腐病提供理论依据。
1 材料与方法
1.1 供试菌株
于2014~2017年,从黑龙江省东部发病较严重两块试验田和生产田采集土壤样品。试验田(SY)分别位于黑龙江省农垦科学院和黑龙江省农科院佳木斯分院,生产田(SC)分别位于佳木斯市桦南县土龙山镇和佳木斯市桦南县曙光农场一队。参考王子迎等叶蝶诱捕法分离土壤中大豆疫霉[15]。参考郝中娜等方法作单游动孢子分离培养[16]。参考刘春来等方法作大豆疫霉分子鉴定[17]。参考Tian等和文景芝等方法测定致病性[18-19]。547个菌株经形态学、分子生物学及致病性鉴定为大豆疫霉,为本试验供试菌株(见表1)。
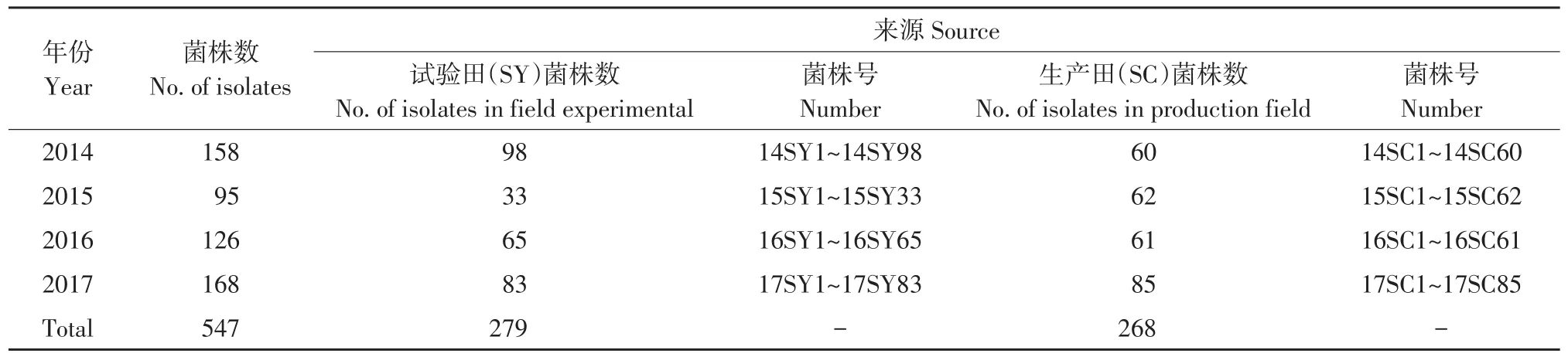
表1 黑龙江省东部大豆疫霉菌株及采集地点Table 1 Phytophthora sojae isolates in eastern Heilongjiang Province and collection sites
1.2 试验方法
参照Frankham CTAB法提取供试菌株DNA[20],提取DNA经1%琼脂糖凝胶电泳检测后,-20℃保存备用。采用徐静静等方法筛选8对SSR引物(见表2)[11],由上海生工生物工程有限公司合成。20 μL PCR扩增体系:10 μL Taq PCR Master Mix(购自上海生工生物工程有限公司),上、下游引物各1 μL,模板DNA 1 μL,无菌去离子水7 μL。反应程序:94℃预变性4 min,94℃变性30 s,40~65℃退火30 s,72℃延伸5 min,40个循环,72℃延伸10 min,4℃保存备用。
SSR引物扩增与银染检测参照杨喆等方法[21]。记录电泳谱带,在电泳图谱上清晰且可重复出现条带记为“1”,相同位置无条带记为“0”,生成“0”和“1”组成原始矩阵。用DPS 7.05中非加权平均法(UPGMA)作聚类分析并建立系统分析树状图。用Population Genetic Analysis 1.3.2分析各SSR标记在各群体内基因频率、有效等位基因数、基因多样性频率及香农信息指数。
2 结果与分析
2.1 大豆疫霉群体遗传多样性分析
2.1.1SSR扩增
8对SSR引物均扩增出多态性条带,多态性比例最高为94.74%,等位基因变异范围11~19。8对引物共扩增出123个条带,其中110个为多态性条带,占比89.43%,表明黑龙江省东部大豆疫霉群体具有丰富遗传多样性。引物PSG172多态性最佳,多态性条带比例为94.74%,引物PSG113多态性较差,多态性条带比例为78.57%。
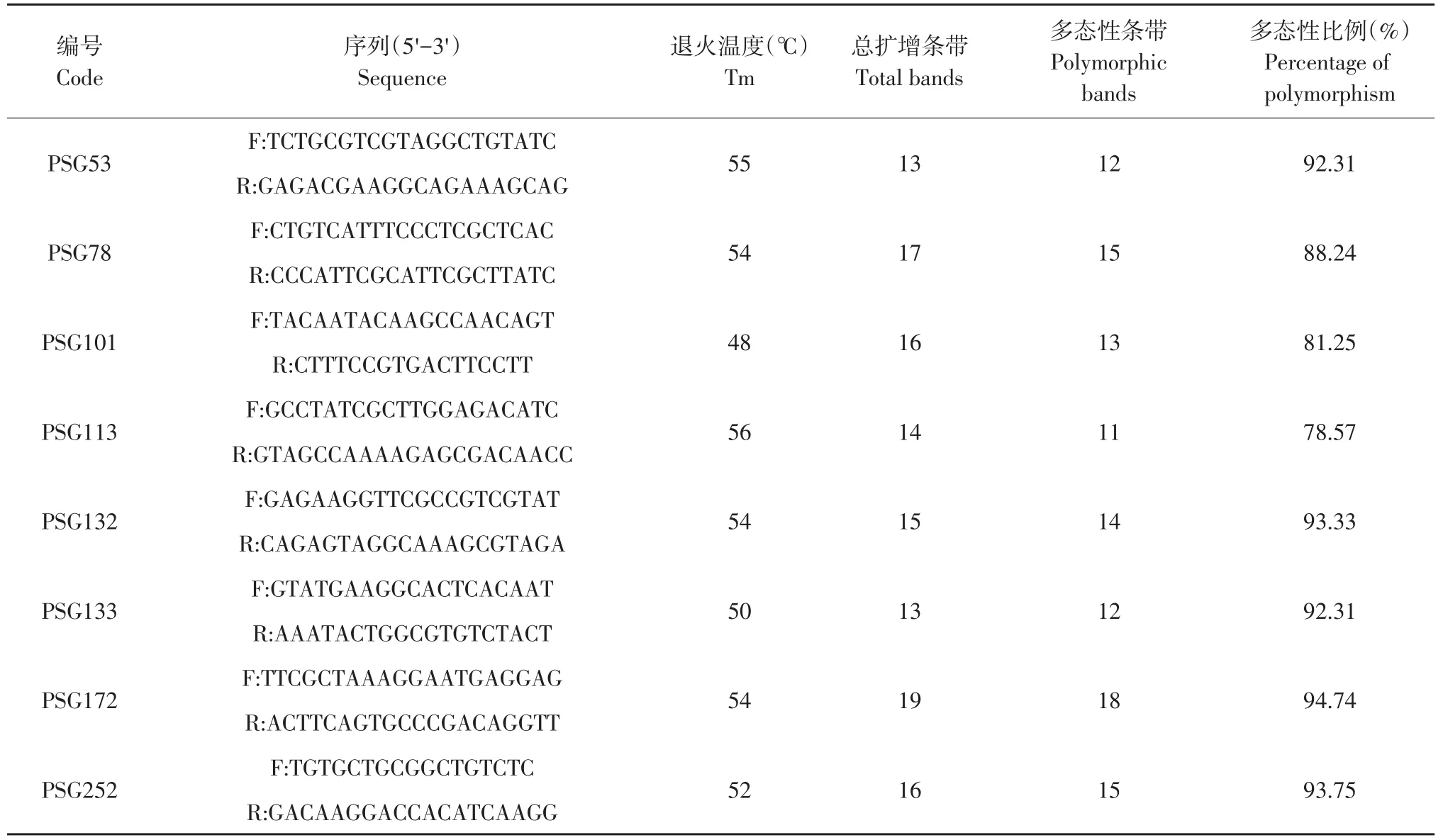
表2 8对引物扩增多态性条带Table 2 Polymorphic bands amplified by eight pairs of primers
2.1.2遗传多样性分析
547个供试菌株基因多态性位点主要分布在700、450和350 bp,其中450 bp基因位点最多,基因频率为0.5265,其次为700 bp和350 bp,基因频率为0.4497;在150~100 bp、<100 bp和100 bp分布基因频率较低,分别为0.1371、0.1517和0.1536(见表3)。
547个供试菌株在450 bp处基因多样性复杂,有效等位基因数为1.9944,基因多样性频率为0.4986,Shannon信息指数为0.6917,其次为700和350 bp,有效等位基因数为1.9800,基因多样性频率分别为0.4949,Shannon信息指数为0.6881。在150~100 bp处基因多样性程度最小,有效等位基因数为1.3100,基因多样性频率为0.2366,Shannon信息指数为0.3997,其次为<100和100 bp,有效等位基因数为1.3476和1.3513,基因多样性频率为0.2574和0.2600,Shannon信息指数为0.4257和0.4288(见表4)。
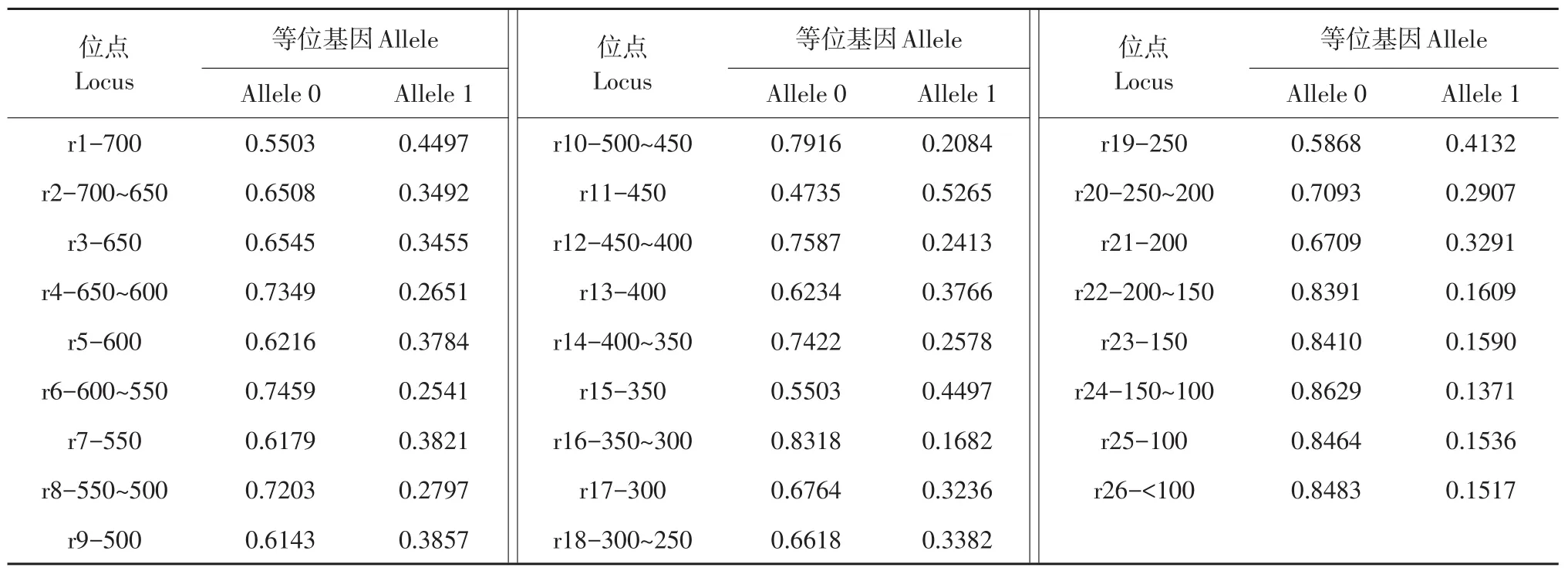
表3 大豆疫霉群体基因频率Table 3 Gene frequency of Phytophthora sojae populations
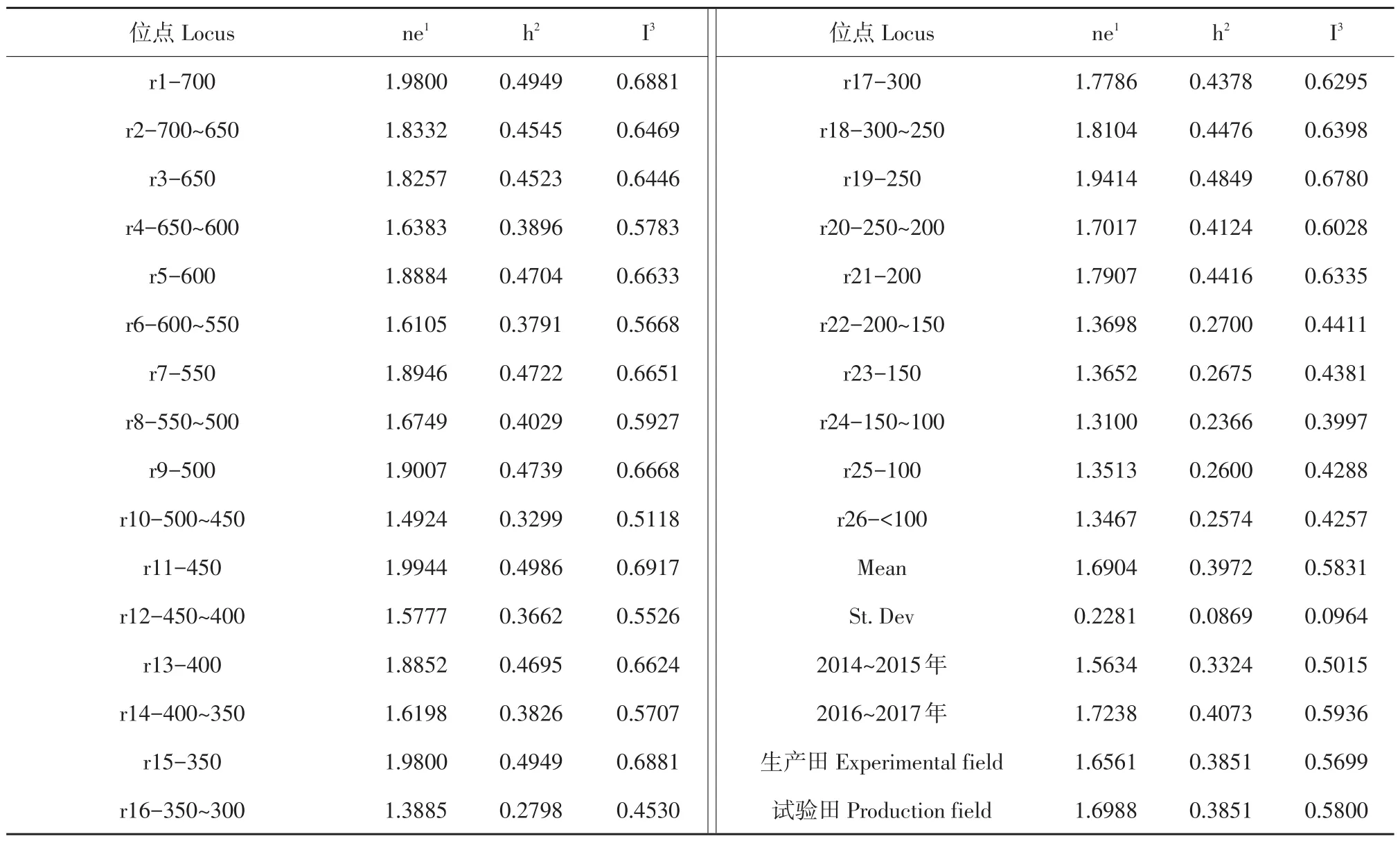
表4 供试菌株遗传多样性参数分析Table 4 Nei's analysis of genetic diversity in subdivided populations
2.2 大豆疫霉群体遗传多样性时间动态
2.2.1遗传多样性时间动态
大豆疫霉群体基因在2014~2015年分布较均匀,仅700~650 bp处基因频率大于0.5,为0.5095。但2016~2017年基因分布分散,在250、450、300、700和350 bp处基因频率均大于0.5,其中250 bp基因频率最高为0.6361,其余基因频率分别为0.5646、0.5476、0.5374和0.5170,波动幅度较大(见表5)。由此可见2016~2017年大豆疫霉群体基因变异程度较大。
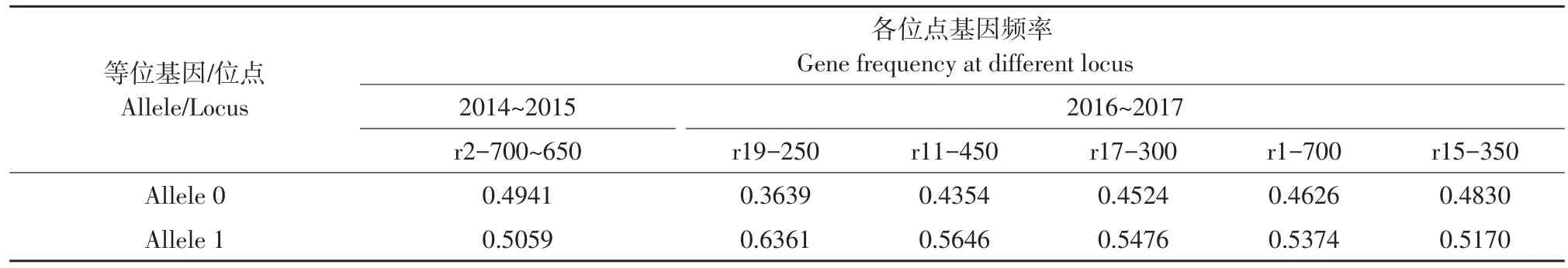
表5 2014~2015年和2016~2017年大豆疫霉群体部分位点基因频率Table 5 Gene frequency of partial Phytophthora sojae populations in 2014~2015 and 2016~2017
与2014~2015年相比,2016~2017年有效等位基因数上升0.1604,基因多样性频率上升0.0749,香农信息指数上升0.0921(见表4)。由表4可知,随时间推移,黑龙江省东部大豆疫霉群体内基因变异极显著,遗传多样性更丰富,基因变异频率提高,说明大豆疫霉群体发生明显进化。
2.2.2聚类分析
当遗传距离为0.8时,聚类分析可将供试菌株划分为5个群组(见表6)。A、B、C和D组中均包含所有年份菌株,E组中仅包含2016~2017年菌株,说明2016~2017年菌株遗传变异程度较高。
2.3 大豆疫霉群体遗传多样性空间动态
2.3.1遗传多样性空间动态
大豆疫霉群体基因在生产田分布较均匀,仅450 bp处基因频率大于0.5,为0.5037,但试验田中600和450 bp处基因频率均大于0.5,其中600 bp基因频率最高为0.5663,450 bp处基因频率为0.5484(见表7)。可见试验田大豆疫霉群体基因变异程度较大。
与生产田相比,试验田群体有效等位基因数上升0.0427,基因多样性频率上升0.0110,香农信息指数上升0.0101(见表4)。表明黑龙江省东部不同地块间大豆疫霉群体基因发生变化,试验田大豆疫霉群体遗传多样性较丰富,基因变异频率略高于生产田。
2.3.2聚类分析
当遗传距离为0.84时,聚类分析可将供试菌株划分为6个群组,A、B、C、D和E组中均包含试验田和生产田菌株,可见不同地块间大豆疫霉群体可能存在基因交流。F组仅包含8个试验田菌株,说明试验田群体遗传多样性略高于生产田群体(见表8)。
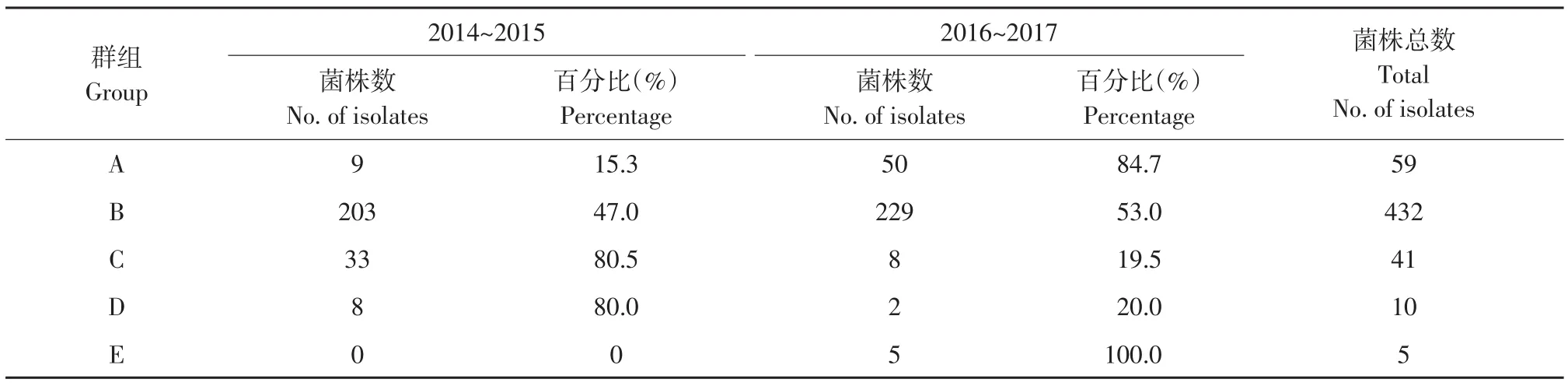
表6 2014~2015年和2016~2017年大豆疫霉群体UPGMA聚类分析结果Table 6 Results of UPGMA Cluster analysis of Phytophthora sojae populations in 2014~2015 and 2016~2017
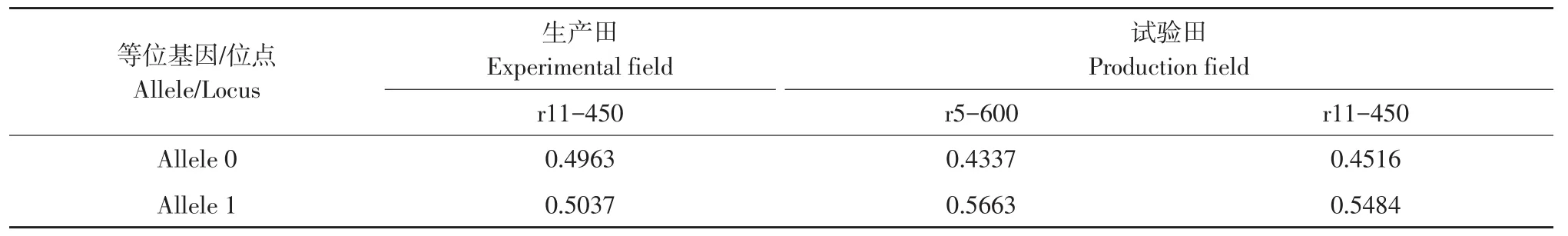
表7 生产田和试验田大豆疫霉群体部分位点基因频率Table 7 Gene frequency of partial Phytophthora sojae populations in production and experimental fields
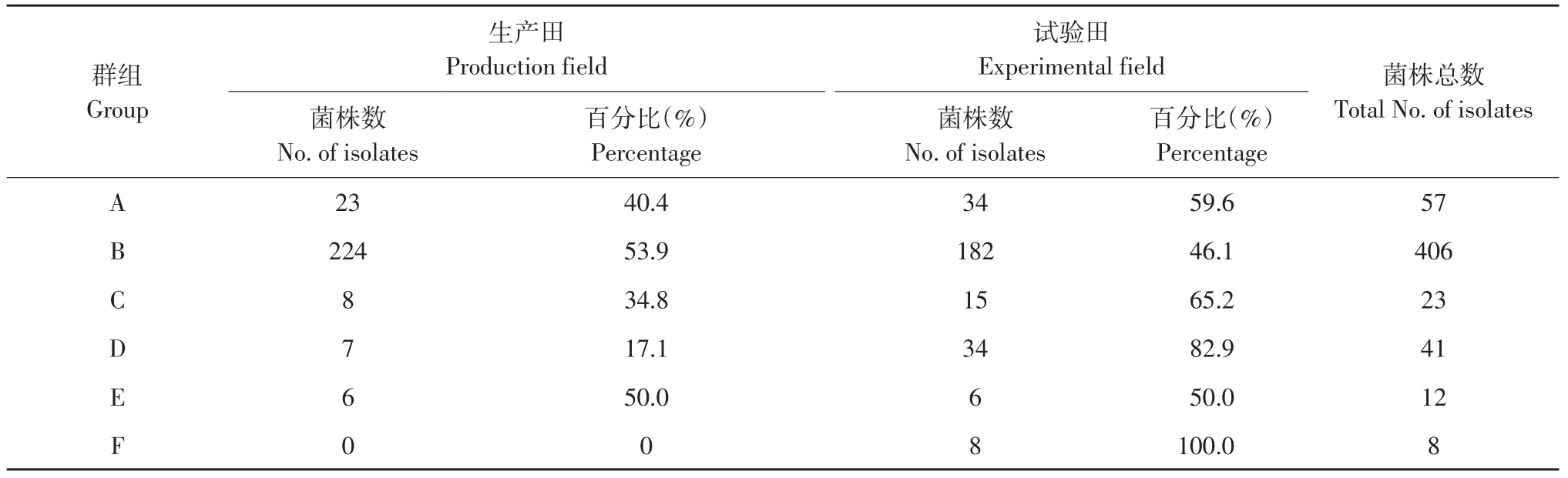
表8 生产田和试验田大豆疫霉群体UPGMA聚类分析结果Table 8 Results of UPGMA cluster analysis of Phytophthora sojae populations in production and experimental fields
3 讨论与结论
病原菌丰富遗传多样性是病害系统中寄主与病原物相互作用结果。更换寄主品种增大对病原物选择压力,病原物种群发生变异,再通过基因交流、有性杂交及基因突变等形式导致种群及遗传结构改变,产生遗传多样性。遗传多样性是评价病原物种群进化潜能及抵御生存压力能力重要指标[20,25]。
为避免大豆品种基因型对土壤中大豆疫霉基因型的选择性偏差,本试验直接从土壤而非病株中分离大豆疫霉。SSR分子标记广泛应用于植物病原菌遗传多样性研究,其分子标记结果反映基因型差异,不受空间和时间限制,可精确揭示群体间遗传多样性。本试验采用8对SSR引物对大豆疫霉均可扩增出多态性条带,多态性比例较高,表明SSR标记引物选择恰当,适用于大豆疫霉遗传多样性研究[11]。
本文首次用SSR分子标记分析大豆疫霉群体时间和空间变化规律,结果显示随时间推移,黑龙江省东部大豆疫霉群体遗传多样性更复杂。Gally等阿根廷大豆疫霉群体RAPD分析结果认为是大豆疫霉群体与寄主长期互作结果[26]。试验田群体遗传多样性略高于生产田群体,但增幅小,说明不同地块间大豆疫霉遗传多样性差异不明显,与殷丽华报道黑龙江省东部不同地区间大豆疫霉群体遗传多样性变化较小结论一致,但本试验采集菌株数更多,结果可信度更高[27]。根据本试验结果推测,未来几年黑龙江东部大豆疫霉群体遗传多样性更加复杂,基因变异频率相应提高。中国不同地区大豆疫霉群体存在广泛遗传变异[28],我国福建省、美国大豆疫霉群体与黑龙江省大豆疫霉群体遗传相似性较低,表明不同地区间大豆疫霉遗传多样性差异较大[29]。本试验发现不同试验田和生产田群体遗传多样性差异不显著,原因可能是供试菌株虽来自不同地块,但整体上仍属于同一地理区域,符合生物进化规律。
本试验采用8对SSR引物分析547株大豆疫霉群体遗传多样性,结果表明,黑龙江省东部大豆疫霉群体具有丰富遗传多样性。随时间推移,大豆疫霉群体发生明显进化,表现在群体内基因发生显著变异,基因变异频率相应提高,遗传多样性更加丰富;试验田大豆疫霉菌群体遗传多样性较丰富,基因变异频率略高于生产田。由于本试验供试大豆疫霉菌株均采自发病严重的黑龙江省东部地区,存在区域局限性,后续应扩大样品采集范围,对各省市大豆疫霉群体作深入研究,监测各地区大豆疫霉群体遗传多样性动态变化,为科学合理使用抗病品种提供依据。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
绵阳师范学院学报(2021年5期)2021-05-28 07:07:46
河北大学学报(自然科学版)(2020年1期)2020-01-15 01:05:22
广东农业科学(2017年5期)2017-08-29 10:37:45
中国医院院长(2017年9期)2017-06-15 12:59:24
知识经济·中国直销(2016年7期)2016-11-07 09:36:18
公民与法治(2016年6期)2016-05-17 04:10:35
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
杭州科技(2014年3期)2014-02-27 15:26:49
