
实时定量PCR测定 爱德华氏菌隐蔽质粒的拷贝数
2018-07-13 09:41:10付立霞韩先干张晓君刘晓丹曾令兵
淡水渔业 2018年4期
付立霞,高 雯,韩先干,高 波,张晓君,刘晓丹,曾令兵
(1.农业部淡水生物多样性保护重点实验室,中国水产科学研究院长江水产研究所,武汉 430223;2.江苏省人畜共患病学重点实验室,扬州大学动物科学与技术学院,江苏扬州 225009;3.中国农业科学院上海兽医研究所,上海 200241)

1 材料和方法
1.1 菌株、质粒和试剂
1.2 引物设计与合成
1.3 质粒和基因组的制备

表1 本研究中所用引物及其序列Tab.1 Primers and its sequences used in this study
注:质粒pEI1中的CDS1(pEI1_CDS1)和pEI2中的CDS1(pEI2_CDS1)为非同源基因,为避免混淆,后续以orf1和orf2代替,以示区分。
1.4 重组质粒的构建
1.4.1质粒pUC18-orf1的构建
1.4.2质粒pUC18-luxS-orf1的构建
1.4.3质粒pUC18-luxS-orf1-orf2的构建
在成功构建质粒pUC18-luxS-orf1的基础上,进一步以引物orf2-F/orf2-R扩增位于隐蔽质粒pEI2上的预测编码基因CDS1,最后通过上述方法将其克隆至质粒pUC18-luxS-orf1的KpnⅠ和EcoRⅠ位点之间,将构建成功的质粒命名为pUC18-luxS-orf1-orf2 (图1),用于后续荧光定量PCR标准曲线的建立。

图1 质粒pUC18-luxS-orf1-orf2的物理图谱Fig.1 Physical map of plasmid pUC18-luxS-orf1-orf2
1.5 荧光定量PCR
荧光定量PCR在ABI 7500荧光定量PCR仪中进行,反应体系为20 μL,其中SYBR Premix Ex Taq Ⅱ 10 μL,上、下游引物及ROX Reference Dye Ⅱ各0.4 μL, DNA模板8.8 μL,每个反应设3个平行重复。反应条件:95 ℃预变性30 s; 95 ℃变性5 s,60 ℃ 复性并延伸34 s,40个循环。随后进行熔解曲线分析以确定引物的特异性,反应程序为:95 ℃ 15 s,60 ℃ 1 min,然后以0.1 ℃/s 的速率升温至95 ℃, 进而形成一个荧光信号随温度升高而减弱的熔解曲线图。
1.6 质粒拷贝数测定标准曲线的的建立
利用超微量分光光度计One Drop OD-1000+测定质粒pUC18-luxS-orf1-orf2浓度,根据下面公式[22]计算其对应拷贝数,然后10倍稀释至108、107、106、105、104拷贝/μL,用作建立标准曲线时荧光定量PCR的反应模板。

反应结束后,以测得Ct值为纵坐标,拷贝数对数为横坐标进行线性回归分析,扩增效率(E)与标准曲线的斜率(k)相关[23],计算公式为:
E=(10-1/k-1)×100%
1.7 质粒拷贝数的计算
同时采用绝对定量和相对定量的方法对质粒拷贝数进行计算,其中绝对定量参照Yu等[24]所述方法进行。相对定量中质粒拷贝数的计算先按照Livak等[25]所述方法判断-ΔΔCt方法对本研究数据处理的适用性,如不适用则参照Pfaffl等[26]所述方法进行质粒拷贝数的计算,公式为:

式中,Etarget为靶基因的扩增效率;Erefere为参考基因的扩增效率;ΔCt target(control-sample)为参照样品中靶基因Ct减去待测试样品中靶基因Ct;ΔCt referet(control-sample)为参照样品中参考基因Ct减去待测试样品中参考基因Ct。
1.8 Geneaid DNA Mini Kit对染色体DNA和质粒DNA的回收率测定
1.9 统计分析
利用Excel 2013进行数据分析,OriginPro 2016软件对不同处理下得到的质粒拷贝数进行单因素方差分析,P<0.05为存在显著性差异,P<0.01为存在极显著性差异,以均值±标准差表示。
2 结果与分析
2.1 重组质粒的构建
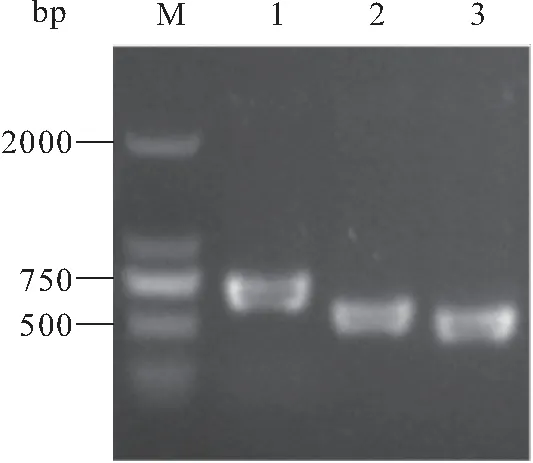
图2 目的基因的PCR扩增Fig.2 PCR amplification of target genesM:DL2000 DNA Marker;1:orf1;2:luxS;3:orf2
对构建成功的质粒通过引物M13-47/ M13-48进行PCR鉴定,可分别扩增出大小约为811 bp,1 310 bp和1 753 bp的预期目的片段(图3),进一步测序结果表明,成功构建质粒pUC18-luxS-orf1-orf2。
2.2 引物特异性评价
引物的特异性通过PCR产物的琼脂糖凝胶电泳和熔解曲线进行分析。电泳结果显示,引物对qluxS-F/qluxS-R,qorf1-F/qorf1-R,qorf2-F/qorf2-R PCR对质粒pUC18-luxS-orf1-orf2和基因组总DNA的扩增产物分别为91 bp,83 bp和130 bp预期大小的单一条带(图4)。熔解曲线亦显示3对引物的PCR产物均表现为单一峰,这表明在分析温度范围内,没有非特异性扩增。

图3 构建质粒的PCR鉴定Fig.3 PCR identification for constructed plasmids M:DNA Marker DL-2000;1:pUC18-orf1;2:pUC18-luxS-orf1;3:pUC18-luxS-orf1-orf2
2.3 标准曲线的建立
在1×104~1×108拷贝/μL浓度范围内,以测得Ct值对模板拷贝数的对数值作图,结果见图5,可见所有曲线均呈线性相关(R2>0.999),斜率分别为-3.285 15、-3.333 5和-3.396、与之相应的扩增效率分别为101.6%、99.5%和 97.0%。荧光定量PCR要求R2>0.99,扩增效率介于90%~110%之间。本研究中,基于3对引物进行的荧光定量PCR均满足条件,表明实验结果可信。

图4 荧光定量PCR产物电泳 Fig.4 Electrophoresis of real-time PCR products M:DL 2000 DNA Marker;1,3,5:以质粒pUC18-luxS-orf1-orf2为模板进行PCR扩增的luxS,orf1和orf2 产物;2,4,6:以 爱德华氏菌总基因组为模板进行PCR扩增的luxS,orf1和orf2产物
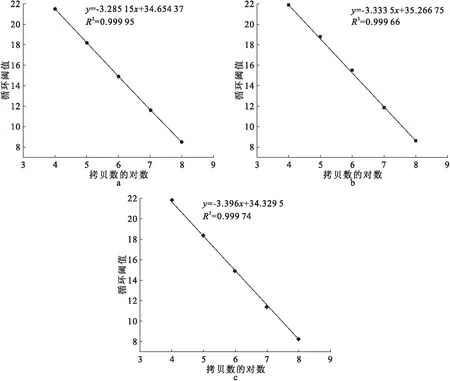
图5 luxS(a)、orf1(b)和orf2(c)标准曲线的建立Fig.5 Construction of the standard curves for luxS (a),orf1 (b) and orf2 (c)
2.4 ΔΔCt方法的适用性评价
ΔΔCt方法计算有效的前提条件为,目的基因和参考基因的扩增效率必须近似相等。为确认这一点,以不同模板稀释度下目的基因与参考基因Ct值的差值ΔCt对模板拷贝数的对数进行绘图,如回归斜率趋近于零即可确认其有效性,而本研究中不同稀释度下,目的基因orf1和orf2相对于参考基因luxS的ΔCt回归斜率分别为-0.034 3(图6a)和-0.145(图6b),这表明ΔΔCt方法适宜于前者而不宜用于后者的质粒拷贝数计算。为保证计算的一致性,后续相对定量根据Pfaffl等[26]所述方法进行。

图6 ΔΔCt计算的有效性Fig.6 Validation of the ΔΔCt calculation for orf1/luxS (a) and orf2/luxS (b)
2.5 质粒拷贝数计算
绝对定量策略下,试剂盒提取的总DNA中,pEI1的拷贝数为3.63±0.30,pEI2的拷贝数为5.04±0.18,而水煮法制备的总DNA中,pEI1和pEI2的拷贝数则分别为11.84±0.80 和11.70±0.25 (表2),两种不同总DNA制备方法下所测得质粒拷贝数差异极显著。

表2 绝对定量测得的质粒拷贝数Tab.2 Estimated plasmid copy number (PCN) by absolute quantification
注:同一列数据上标中不同大写字母表示差异极显著(P<0.01),下同。
相对定量策略下,试剂盒提取的总DNA中,pEI1和pEI2的拷贝数分别为4.22±0.15和5.13±0.50,而水煮法制备的总DNA中,pEI1和pEI2的拷贝数则分别为13.85±1.64 和11.90±0.97(表3),两种不同总DNA制备方法下所测得质粒拷贝数差异极显著。

表3 相对定量测得的质粒拷贝数Tab.3 Estimated plasmid copy number (PCN) by relative quantification
2.6 Geneaid DNA Mini Kit对染色体DNA和质粒DNA的回收率
3 讨论
在利用实时荧光定量PCR进行质粒拷贝数测定时,主要有两种定量策略:绝对定量和相对定量,绝对定量需构建标准品及标准曲线,据此来推算未知样本的量;而相对定量则只需确定样本中靶序列相对于另一参照样本的量的变化即可,根据不同的计算方法又可分为2-ΔΔCt法和Pfaffl法等[25,26],其中前者要求目的基因与内参基因扩增效率基本相等且接近于100%,在实验中通常难以得到满足,而后者则充分考虑了扩增效率对结果的影响,更具有可行性,结果也更为准确。在Lee等[27]对大肠杆菌质粒拷贝数的测定中,基于2-ΔΔCt法的相对定量与绝对定量的计算结果基本一致。本研究中,由于针对luxS、orf1和orf2的引物扩增效率不完全一致,分别为101.6%、99.5%、97.0%,最终采用Pfaffl法进行相对定量,计算结果显示,除水煮法组的pEI1拷贝数在两种定量策略下存在差异之外(分别为11.84±0.80和13.85±1.64),其余皆无显著差异,这表明只要选择合适的相对定量方法,其计算结果与绝对定量策略下的计算结果具有一致性。
猜你喜欢
河北医学(2021年10期)2021-10-27 00:37:14
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
中成药(2018年12期)2018-12-29 12:25:44
食品科学(2018年10期)2018-05-23 01:27:28
中成药(2017年6期)2017-06-13 07:30:35
西南医科大学学报(2015年1期)2015-08-22 13:01:46
医学研究杂志(2015年4期)2015-06-10 06:42:43
中国当代医药(2015年9期)2015-03-01 02:01:59
发明与创新(2015年25期)2015-02-27 10:39:16
