
CRISPR/Cas技术及其在发酵菌株上的应用
2018-07-04 05:55:04石宏武崔晟榕马小军
微生物学杂志 2018年2期
石宏武, 乔 晶, 崔晟榕, 马小军,2*
(1.中国医学科学院北京协和医学院 药用植物研究所,北京 100193;2.中国医学科学院药用植物研究所 云南分所,云南 景洪 666100)
在工业发酵生产上,利用发酵菌株的定向代谢途径,可将低碳链化合物(如葡萄糖、氨基酸、甘油等)转化成具有重要价值的高碳骨架化合物(如萜类、黄酮类、甾醇等),而获得优良的发酵菌株一直是发酵工业上的一项重要任务。在发酵菌株的改良方法上,通常是通过人工长期驯化,或基因突变(如射线处理),或是通过转基因技术(如农杆菌介导、电转化法、基因枪法等),将外源基因导入受体细胞中,再通过受体细胞对外源基因整合,使其稳定遗传表达[1]。这些方法存在很多不足,如人工驯化周期长、成本高;或是存在外源基因的随机性整合对受体基因组信息造成意外损坏,受体对外源基因的整合效率低,整合的片段易丢失等问题[2]。为应对工业发酵上菌株低效率改良的问题,基因靶向编辑技术(gene targeted editing technology)应运而生。其中,规律成簇间隔短回文重复Cas(Clustered Regularly Interspaced Short Palindromic Repeats/Cas,CRISPR/Cas)是基于细菌及古生菌中的适应性免疫系统而改造的一项基因编辑技术,可以实现对单个或多个目的基因进行指定位点编辑,该项技术不仅大幅度提高了基因的转化水平,还减少了对受体细胞的基因组信息的随机性破坏,使其快速构建稳定高产的发酵菌株和精确优化发酵工艺条件等成为可能。
1 CRISPR/Cas系统
CRISPR/Cas系统早期是在大肠埃希菌中发现这种特定的重复间隔序列结构,生物信息学分析表明,这种特殊的重复间隔序列与外源的噬菌体或质粒中某些特定序列具有高度的相似性,推测其功能与抵御外源遗传物质入侵有关,并随后在嗜热链球菌(Streptococcusthermophilus)的免疫试验中证实了这一猜想[3-4]。
1.1 CRISPR/Cas系统的基本结构
CRISPR/Cas系统由高度保守的间隔重复序列(CRISPR)和Cas酶基因组成(图1)。在CRISPR序列中,具有免疫“记忆”功能的重复序列(Repeats)和间隔序列(Spacers)依次排列[5]。Cas基因则是位于CRISPR前端附近的一段保守序列,其编码的蛋白酶具有核酸酶和切口酶活性,能对DNA进行剪切和修复。此外,在CRISPR位点与Cas序列之间存在一个前导序列(Leader sequence),负责启动CRISPR序列的转录。这些单位结构间地协调工作,保证了细菌或古生菌免受外源病毒的侵扰[3]。
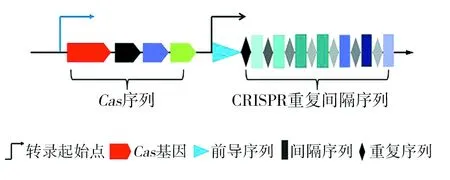
图1 CRISPR和Cas的序列结构Fig.1 The sequence structure of CRISPR and Cas
1.2 CRISPR/Cas系统的工作原理
CRISPR/Cas系统的免疫作用主要分为三阶段:获取、表达和干扰(图2[6])。在获取阶段,当外源噬菌体或质粒入侵时,宿主细胞中的CRISPR/Cas系统首先识别入侵的DNA中原型间隔序列(Protospacers)和原型间隔序列毗邻序列(Protospacer adjacent motif,PAM)位点,产生的Cas酶复合物(主要为Cas1和Cas2)对原型间隔序列进行裂解并整合到宿主细胞的CRISPR序列中的第一个重复序列和前导序列之间,形成“记忆”片段[7]。在表达阶段,CRISPR序列在RNA聚合酶作用下转录成一段CRISPR RNA前体pre-crRNA,接着pre-crRNA在核糖核酸内切酶的剪切下形成较为成熟的CRISPR crRNA[8]。在干扰阶段,成熟的CRISPR crRNA结合Cas蛋白复合物,对入侵DNA上具有的PAM进行识别,Cas蛋白复合物对PAM位点上的上游3~8 bp位置进行剪切,从而发挥免疫作用[9]。

图2 II型CRISPR/Cas系统的工作原理图[6]Fig.2 The working principle diagram of type II CRISPR/Cas system[6]
1.3 CRISPR/Cas系统的分类
根据Cas基因的保守性、表达的核心元件序列以及整合系统的不同,CRISPR/Cas系统分为3种类型:Type I、Type II、Type III[10]。Type I和Type III在生化结构上具有相似的特点,即由多种特殊的Cas蛋白构成的CASCADE复合物。但在Type I中,多亚基的CASCADE结合pre-crRNA,在Cas6的剪切下,在5′端具有8个延长的核苷酸,3′端由间隔序列和部分重复序列形成发卡结构,crRNA结合CASCADE,在Cas3亚族酶协助下通过PAM识别互补的DNA靶位点,进行剪切[11]。而在Type III中,crRNA的形成需要Cas6,而在干扰阶段的识别中不一定需要PAM[12]。Type II中的Cas序列则主要由编码四种不同蛋白酶基因组成[8](图3[6]),其中Cas9基因是参与免疫防御的标记基因,其转录的蛋白是由1 409个氨基酸组成,含有两个核酸酶结构域:HNH结构域和氨基端的Ruvc-like结构域,HNH结构域可切割与crRNA互补配对的模板链,Ruvc-like结构域切割对应的另一条模板链[13]。在Type II工作中, pre-crRNA序列经过核糖核酸酶RNase III剪切形成成熟的crRNA,接着与反式激活crRNA(tracrRNA)进行碱基互补配对,形成嵌合的sgRNA,sgRNA与Cas9结合,通过PAM识别靶基因并进行剪切[14]。研究还发现,三种免疫系统的分布也不均一,Type I主要存在于细菌和古生菌中,Type II存在于少数的古生菌和大部分的细菌中,而Type III通常只在古生菌中出现[15]。
2 基于CRISPR/Cas系统的基因编辑技术
DNA双链断裂产生的信号(Double Strand Break,DSB)会激活细胞内固有的非同源末端连接(Non-homologous ending-joining,NHEJ)或同源重组(Homology directed recombination repair,HDR)两种不同的修复机制,从而对损伤的DNA序列进行修复[16](图4[17]),其中,NHEJ修复能使碱基片段缺失或插入,HDR修复可实现外源基因或大片段的整合。
CRISPR/Cas系统能够利用Cas酶对DNA进行切割,产生DSB信号从而激活DNA损伤修复机制。要想利用CRISPR/Cas技术对基因进行定点编辑,就需要对tracrRNA-crRNA复合体进行改造,设计出能够被Cas蛋白识别并引导其结合靶位点的sgRNA。CRISPR/Cas技术可实现特定位点的基因敲入、敲除,相比传统的基因靶向技术,其更为精确、高效且经济,为生命科学研究提供了有利的技术支持。CRISPR/Cas技术的使用一般需要经历以下六个步骤[18]:首先是结合PAM结构筛选靶向基因的识别位点,接着设计合成sgRNA,然后根据sgRNA序列模板合成一对序列互补的DNA双链,接着与带有报告基因的Cas载体连接,体外检测构建的sgRNA的活性,然后将构建的载体转染细胞并通过测序进行结果验证。其中,设计构建sgRNA是关键步骤,能否设计出高活性、高精准性的sgRNA关系着该技术实施的成败。

图3 三种类型的CRISPR/Cas系统中的Cas蛋白组成[6]Fig.3 Cas proteins in Type I, II, and III CRISPR-Cas systems[6]
cas1和cas2为普通蛋白;cas3、cas9和cas10为标记蛋白;其余为特征蛋白
cas1 and cas2 are universally proteins; cas3,cas9 and cas10 are Signature proteins;the rest are type-dependentproteins
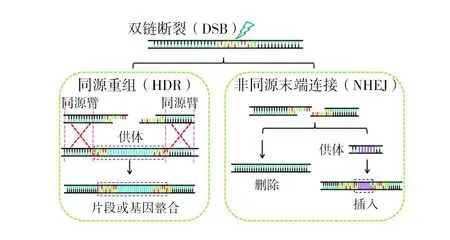
图4 由DSB信号引起的DNA自身修复[17]Fig.4 DNA self repair induced by DSB signa[17]
3 CRISPR/Cas技术在发酵菌株上的研究应用
随着多物种基因组测序工作的完成和合成生物学的发展,利用发酵菌株的代谢途径经过发酵生产食品、医药及化工等原料是未来发展的趋势,且很多植物次生代谢产物已经在发酵菌株中实现了快速合成,这些使得传统的微生物发酵工程逐渐上升到一个新阶段。目前的研究报道中,采用CRISPR/Cas基因编辑技术在发酵菌株上的应用已经取得了较好的成效,其中在细菌、酵母和曲霉等发酵菌株上的研究尤为引人注目。
3.1 在细菌发酵上的研究应用
在细菌发酵上常用的发酵菌株有大肠埃希菌、醋酸杆菌、乳酸杆菌、枯草杆菌、丙酮丁醇梭杆菌和谷氨酸棒状杆菌等。在发酵工程上,通过对这些细菌微生物的代谢通路定向改造和驯化,可获得高产的工业发酵菌株。CRISPR/Cas技术对菌株基因组的定点编辑,能够快速获得特异性菌株,大幅度提高发酵菌株的改良优化效率,节约时间成本。在基础研究构建特定缺陷型菌株上,与通常的物理、化学突变方法相比,CRISPR/Cas技术目的更明确,成功率更高。此外,CRISPR/Cas技术还能解决以往出现的由于质粒丢失导致的目标途径受阻、增强发酵菌株抵抗病毒的能力等棘手问题。
在发酵工业上,若大罐液体发酵受到噬菌体严重污染,轻则引起发酵周期延长、发酵液变清和发酵产物难以形成等事故,重则造成倒罐、停产甚至危及工厂命运。为提高枯草芽胞杆菌(Bacillussubtilis)对噬菌体SPP1的抵抗性,Lina等[19]克隆噬热链球菌(S.thermophilus)中的Cas9基因,并构建了针对SPP1噬菌体的间隔序列,B.subtilis被导入构建的载体后,显示其对噬菌体SPP1的感染具有较好的免疫性。该研究为工业发酵中解决噬菌体污染问题提供了新思路。
细菌微生物发酵生产清洁环保的生物能源,降低了工业生产对化石能源的依赖性,提高了天然纤维素的有效使用率。Wen等[20]提出的整合生物发酵工艺,将梭杆菌属的Clostridiumcellulovorans和Clostridiumbeijerinckii联合构建成成对梭状芽胞杆菌(twin-clostridial consortium),并进一步敲除C.cellulovorans中的乙酸激酶基因(Clocel_1892)和乳酸脱氢酶基因(Clocel_1533),过表达丁酸激酶基因(Clocel_3674),与此同时,采用CRISPR技术下调了氢化酶基因(Clocel_2243)的表达,在C.beijerinckii上设计了有机酸同化和戊糖途径相关基因,最终使得构建的成对菌株在分解发酵83.2 g/L的木质纤维素后,可产生22.1 g/L的溶媒(包含4.25 g/L的丙酮,11.5 g/L的丁醇和6.37 g/L的乙醇)。Wang等[21]在ClostridiumsaccharoperbutylacetonicumN1-4上采用CCRISPR技术对编码乙酸和丁酸的必要基因pta、buk进行敲除,然后以启动子PJ23119优化,获得的菌株在P2培养基上经过发酵可产生19.0 g/L的丁醇。Mark等[22]利用SpCRISPR-Cas9技术,转录抑制丙酮丁醇梭杆菌(Clostridiumacetobutylicum)中编码HPr激酶/磷酸化酶基因,靶向控制碳代谢产物走向,使来自木质纤维素中的葡萄糖和木糖被很好地利用。CRISPR/Cas技术对发酵菌株的快速定向改造,为工业化大量生产清洁能源提供了便利,也大大提高了发酵原料的有效使用率。
大肠埃希菌基于其较清晰的遗传背景、完善的载体受体系统和生长迅速等优势,使其在发酵生产上广为应用。Li等[23]研究了大肠埃希菌(Escherichiacoli)中β-胡萝卜素合成相关基因修饰对其产量的影响,采用CRISPR/Cas技术构建33种修饰的100个组合方式的基因突变体菌株,最终筛选到较好的15种突变体,间歇发酵β-胡萝卜素产量达2.0 g/L。同样地,Xia等[24]利用一个二元载体构建的CRISPR/Cas偶联λ-Red重组酶系统,PCR扩增构建来自Shewanellafrigidimarina的fadR的转录因子以及乙酰-CoA羧化酶基因,经过电转化方式对大肠埃希菌脂肪酸代谢相关基因进行快速编辑,获得两种大肠埃希菌突变体,突变体的脂肪酸含量比野生型分别高13.1%和5.3%。此外,研究发现乙酰-CoA和丙二酰-CoA的含量决定了大肠埃希菌合成生松素和柚皮素的发酵产率。Wu等[25]利用CRISPR/Cas技术对乙酰-CoA和丙二酰-CoA两者代谢支路(如三羧酸循环、糖酵解以及脂肪酸合成途径等)上的相关酶基因进行抑制,使得目标产物生松素和柚皮素的产量分别提高到260.1 mg/L和421.6 mg/L。该团队还通过利用CRISPR技术下调大肠埃希菌脂肪酸生物合成途径中的fabD,fabH,fabB,fabF,fabI关键基因,优化后生产白藜芦醇的质量浓度可达304.5 mg/L[26]。大肠埃希菌的快速繁殖能力为生产医药类化合物提供便利,在一定程度上保证了医药化工原材料的供应。
此外,利用CRISPR技术对发酵细菌菌株代谢途径加以改造,发酵生产脂肪酸酯聚合物[27]、氨基酸[28]、正丁醇[29]和异丙醇[30]等都取得了较好效果,这些基础研究工作为工业发酵构建适宜的发酵菌株提供了参考,为发酵生产中菌株改进提供便捷。基于该技术在细菌发酵上的逐步成熟,未来发酵工业将会迈向一个更高的台阶。
3.2 在酵母发酵上的研究应用
酵母菌株是实验室基础研究和工业生产上重要的一类微生物,可以作为次生代谢、信号转导和物质转运等研究模型,还可以通过发酵大量生产各种化合物。利用CRISPR技术对酵母进行改造,可使其更好地为生命科学服务。

利用CRISPR技术不仅可以提高酵母发酵生产的效率,还可以改善酵母本身存在的缺陷,使其具备特有的性质。酵母发酵会产生一种潜在的致癌物质氨基甲酸乙酯,Chin等[38]采用CRISPR/Cas9对氨基甲酸乙酯合成的关键基因CAR1进行敲除,结果氨基甲酸乙酯和尿素含量大大降低。酵母发酵菌株筛选中出现的抗生素抵抗问题使安全发酵受到威胁,Tsai等[39]构建的大肠埃希菌SR8去除抗生素抗性基因的表达,大大提高了大规模发酵中的安全性。此外,在基础研究上,使用CRISPR/Cas9系统对S.cerevisiae的can1基因进行编辑,敲入一条90 bp的外源双链DNA,可以获得具有刀豆氨酸抗性的特异性酵母突变体[40]。这些对酵母菌株的改造,使之具备特有的性质,为科学研究和工业化生产带来了切实的便利,CRISPR技术的应用,使酵母菌株的研究和应用变得更加深入。
3.3 在曲霉发酵上的研究应用
常用的发酵曲霉属菌株有黑曲霉(Aspergillusniger)和米曲霉(Aspergillusoryzae),黑曲霉是酿酒制醋的主要菌种,米曲霉是传统酿造食品酱和酱油的生产菌种,两种曲霉皆可用于生产淀粉酶、蛋白酶和曲酸等。现代曲霉发酵工艺通常用其生产各种酶制剂、有机酸和农业上的糖化饲料等。
利用CRISPR技术对发酵曲霉进行改造,可改善其发酵生产率,提高发酵原料的有效使用率。在生化研究中使用的半乳糖二酸,可通过果胶中D-半乳糖醛酸氧化生成。Kuivanen等[41]通过转录组学分析半乳糖二酸的分解代谢途径,利用CRISPR/Cas9敲除了A.niger中半乳糖二酸代谢相关的基因,过表达糖醛酸脱氢酶,从而有利于半乳糖二酸的生成,综合了果胶生成半乳糖二酸的生物处理过程。利用米曲霉生产酶制剂和异源蛋白上,Katayama等[42]以CRISPR/Cas9技术靶向编辑A.oryzae,从而获得了所期待的表型转化菌株,为CRISPR/Cas技术应用于米曲霉发酵提供了参考。
4 CRISPR/Cas技术存在的问题及改进
4.1 在发酵菌株中的不足及改进
由于裂殖酵母(Schizosaccharomycespombe)不能合成特定的sgRNA序列,使得CRISPR/Cas基因编辑技术的使用受到限制。研究表明,采用rrk1的RNA启动子和核糖体酶来合成gRNA的方法,可以解除CRISPR/Cas9技术在裂殖酵母(S.pombe)上应用的限制[43]。此外,在体内外合成gRNA采用的U6和U3 snRNA启动子在转录上具有局限性,Gao等[44]设计的一种RGR基因,转录生成的初级的gRNA两端具有核酸酶序列,能够自我催化剪切形成gRNA,RGR基因的转录不但不再受到启动子的限制,而且可以通过通用引物有效地检测产生的突变。这些限制的打破,使得CRISPR/Cas技术在发酵菌株上变得更实用。
4.2 自身存在的脱靶问题及优化
在CRISPR/Cas技术中,由于成熟的gRNA引导Cas蛋白酶只对具有PAM靶位点的上游3~8 bp位置进行剪切,也就是说,PAM上的NGG结构具有的局限性致使gRNA脱靶或是由于其错配而引起的非靶向剪切[45]。此外,在靶向DNA或是gRNA上存在凸起结构,也会引起脱靶效应[46]。所以,CRISPR/Cas基因靶向编辑技术和以往研究的ZFN、TALEN一样,都会面临最致命的脱靶问题[47-48]。
为减少CRISPR/Cas9系统在基因编辑过程中的因错配剪切而造成细胞毒性,通过对来自嗜热链球菌(S.thermophilus)CRISPR/Cas的研究发现,适宜的tracrRNA的最小长度、环状结构、II型结合区和错配的低耐受性,可使StCas9系统在酵母细胞中的剪切准确率提高12%[49]。此外,在探索适宜的sgRNA长度以提高CRISPR/Cas系统的特异性时,发现与目标DNA互补的sgRNA长度为17~18 nt时的特异性更强[50]。同样,通过缩短gRNA 3′端和在5′端加上两个鸟嘌呤核苷酸,也能降低脱靶率[51]。另外,与以往基因编辑技术(ZFNs和TALEN)的脱靶研究中发现的相同,单聚体的稳定性比二聚体要差,为此,通过设计采用两个Cas9核酸酶分别与两条gRNA结合成二聚体结构,发现与对照相比,稳定性可提高50倍以上[52]。这种同时运用两个gRNA进行靶位导向,使得CRISPR/Cas系统对DNA的切割更为严谨。除了对系统序列结构上的改进外,研究还发现Cas9核酸酶的浓度对靶向性剪切也有很大影响,高浓度、低活性的Cas酶能够较好地提高剪切的准确率[45]。
5 展 望
目前,通过微生物发酵工业生产医药、化工等原料已经成为热点。发酵工业生产所需要的发酵菌株大多是通过经典的转基因手段来获得的,而这种菌株在经过几代培养后,经常会出现主要性状质粒丢失的问题,而通过基因诱变筛选的方法往往较难获得指定的缺陷型菌株,对诱变菌株遗传背景也不是很清楚。CRISPR/Cas技术的出现,使得定向构建菌株成为可能,此外,还可以更快更准确地对目标代谢途径中的相关基因互相影响进行研究,以制定高产的优化方案,提高工业化发酵的生产效率。
无论是从发酵工程系统结构上进行优化,还是对Cas酶剪切效率的评估,都是将CRISPR/Cas技术从基础研究向广泛应用推进了一大步。但是,CRISPR/Cas系统中的Type I、Type II、Type III的编辑机理还需进一步研究,特别是Type III是如何在不需要PAM下进行精确地靶向剪切。此外,由于PAM在定位中的作用,使得在寻找DNA靶点时具有一定的限制性。最值得关注的是,CRISPR/Cas在进行基因编辑后,其对机体的后期影响如何,是否对基因遗传表达产生不利的影响,这些问题还亟待进一步研究来阐明。
虽然CRISPR/Cas技术存在的脱靶效应以及特异性等问题,会在一定程度上限制其推广应用,但是随着科研人员对该技术研究的不断深入,使其稳定性与广泛应用性进一步完善。美国国立卫生研究院(NIH)顾问委员会已批准利用CRISPR/Cas9强化依赖于患者T细胞的癌症疗法的申请,这也预示着该技术的成熟并开始走向商业化,相信在未来几年里,成熟的CRISPR/Cas技术在发酵工业领域将广泛应用。
参考文献:
[1] Steensels J, Snoek T, Meersman E,et al.Improving industrial yeast strains:exploiting naturaland artificial diversity[J].Fems Microbiol Rev,2014,38(5):947-995.
[2] 王俊明.利用重组大肠杆菌和核糖开关进行L-赖氨酸生产的研究[D].济南:山东大学,2015.
[3] Pourcel C, Salvignol G, Vergnaud G.CRISPR elements inYersiniapestisacquire new repeats by preferential uptake of bacteriophage DNA,and provide additional tools for evolutionary studies[J].Microbiology,2005,151(3):653-663.
[4] Wiedenheft B,Sternberg SH,Doudna JA.RNA-guided genetic silencing systems in bacteria and archaea[J].Nature,2012,482(7385):331-338.
[5] Horvath P,Barrangou R.CRISPR/Cas,the immune system of bacteria and archaea[J].Science,2010,327(5962):167-170.
[6] Bhaya D,Davison M,Barrangou R.CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation[J].Annu Rev Genet,2011,45(45):273-297.
[7] Liu L,Fan XD.CRISPR-Cas system: a powerful tool for genome engineering[J].Plant Mol Biol,2014,85(3):209-218.
[8] Wright AV,Nunez JK,Doudna JA.Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering[J].Cell,2016,164(1-2):29-44.
[9] Barrangou R.Cas9 targeting and the CRISPR revolution [J].Science,2014,344(6185):707-708.
[10] Makarova KS,Haft DH,Barrangou R,et al.Evolution and classification of the CRISPR-Cas systems[J].Nat Rev Microbiol,2011,9(6):467-477.
[11] Carte J,Pfister NT,Compton MM,et al.Binding and cleavage of CRISPR RNA by Cas6[J].Rna,2010,16(11):2181-2188.
[12] Sontheimer E J,Marraffini L A.Microbiology: slicer for DNA[J].Nature,2010,468(7320):45-46.
[13] Makarova KS,Grishin NV,Shabalina SA,et al.A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action[J].Biol direct,2006,1(1):7, doi: 10.1186/1745-6150-1-7.
[14] Deltcheva E,Chylinski K,Sharma CM,et al.CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III[J].Nature,2011,471(7340):602-607.
[15] Burstein D,Harrington LB,Strutt SC,et al.New CRISPR-Cas systems from uncultivated microbes[J].Nature,2016,542(7640):237-241.
[16] Elcin U.Guardian of genome integrity: cohesin and DNA damage repair[M].United Kingdom:VDM Verlag Dr. Müller,2011:5-93.
[17] Joung JK,Sander JD.TALENs: a widely applicable technology for targeted genome editing[J].Nat Rev Mol Cell Bio,2013,14(1):49-55.
[18] 姜泽群,杨烨.CRISPR/Cas技术在生物医药研究中的应用[J].药物生物技术,2016,23(5):412-416.
[19] Lina JG,Giedrius G.Design of a CRISPR-Cas system to increase resistance ofBacillussubtilisto bacteriophage SPP1[J].J Ind Microbiol Biotechnol,2016,43(8):1183-1188.
[20] Wen Z,Minton NP,Zhang Y,et al.Enhanced solvent production by metabolic engineering of a twin-clostridial consortium[J].Metab Eng,2017,39:38-48.
[21] Wang S,Dong S,Wang P,et al.Genome editing inClostridiumsaccharoperbutylacetonicumN1-4 using CRISPR-Cas9 system[J].Appl Environ Microbiol,2017,doi:10.1128/AEM.00233-17.
[22] Mark RB,Michael EP,Murray MY,et al.Extending CRISPR-Cas9 technology from genome editing to transcriptional engineering in the genusClostridium[J].Appl Environ Microbiol,2016,82(20):6109-6119.
[23] Li Y,Lin Z,Huang C,et al.Metabolic engineering ofEscherichiacoliusingCRISPR-Cas9 meditated genome editing[J].Metab Eng,2015,31:13-21.
[24] Xia J,Wang L,Zhu JB,et al.Expression ofShewanellafrigidimarinafatty acid metabolic genes inE.coliby CRISPR/cas9-coupled lambda red recombineering[J].Biotechnol Lett,2016,38(1):117-122.
[25] Wu J,Du G,Chen J,et al.Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system inEscherichiacoli[J].Sci Rep,2015,5:13477.
[26] Wu J,Zhou P,Zhang X,et al.Efficient de novo synthesis of resveratrol by metabolically engineeredEscherichiacoli[J].J Ind Microbiol Biotechnol,2017,44(7):1-13.
[27] Li L,Ren YL,Chen JC,et al.Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: Controllable P(3HB-co-4HB) biosynthesis[J].Metab Eng,2015,29:160-168.
[28] Cleto S,Jensen JV,Wendisch VF,et al.Corynebacteriumglutamicummetabolic engineering with CRISPR interference (CRISPRi)[J].ACS Synth Biol,2016,5(5):375-385.
[29] Heo MJ,Jung HM,Um J,et al.Controlling citrate synthase expression by CRISPR/Cas9 genome editing forn-butanol production inEscherichiacoli[J].ACS Synth Biol,2017,6(2):182-189.
[30] Liang L,Liu R,Garst AD,et al.CRISPR EnAbled Trackable genome Engineering for isopropanol production inEscherichiacoli[J].Metab Eng,2017,41:1-10.
[32] 张根林.酿酒酵母中β-香树脂醇合成途径的构建与调控[D].北京:北京理工大学,2015.
[33] Arendt P,Miettinen K,Pollier J,et al.An endoplasmic reticulum-engineered yeast platform for overproduction of triterpenoids[J].Metab Eng,2017,40:165-175.
[34] Amanda AR,Leo D,Maren W,et al.A Cas9-based toolkit to program gene expression inSaccharomycescerevisiae[J].Nucleic Acids Res,2017,45(1):496-508.
[35] Lee YG,Jin YS,Cha YL,et al.Bioethanol production from cellulosic hydrolysates by engineered industrialSaccharomycescerevisiae[J].Bioresour Technol,2017,228:355-361.
[36] Lian JZ,Zhao H.Construction of plasmids with tunable copy numbers inSaccharomycescerevisiaeand their applications in pathway optimization and multiplex genome integration[J].Biotechnol Bioeng,2016,113(11):2462-2473.
[37] Klein M,Carrillo M,Xiberras J,et al.Towards the exploitation of glycerol′s high reducing power inSaccharomycescerevisiae-based bioprocesses[J].Metab Eng,2016,38:464-472.
[38] Chin YW,Kang WK,Jang HW,et al.CAR1 deletion by CRISPR/Cas9 reduces formation of ethyl carbamate from ethanol fermentation bySaccharomycescerevisiae[J].J Ind Microbiol Biotechnol,2016,43(11):1517-1525.
[39] Tsai CS,Kong II,Lesmana A,et al.Rapid and marker-free refactoring of xylose-fermenting yeast strains with Cas9/CRISPR[J].Biotechnol Bioeng,2015,112(11):2406-2411.
[40] Dicarlo JE,Norville JE,Mali P,et al.Genome engineering inSaccharomycescerevisiaeusing CRISPR-Cas systems[J].Nucleic Acids Res,2013,41(7):4336-4343.
[41] Kuivanen J,Wang YJ,Richard P.EngineeringAspergillusnigerfor galactaric acid production: elimination of galactaric acid catabolism by using RNA sequencing and CRISPR/Cas9[J].Microb Cell Fact,2016,15(1):210,doi:10.1186/s12934-016-0613-5.
[42] Katayama T,Tanaka Y,Okabe T,et al.Development of a genome editing technique using the CRISPR/Cas9 system in the industrial filamentous fungusAspergillusoryzae[J].Biotechnol Lett,2016,38(4):637-642.
[43] Jacobs JZ,Ciccaglione KM,Tournier V,et al.Implementation of the CRISPR-Cas9 system in fission yeast[J].Nat Commun,2014,5:5344,doi: 10.1038/ncomms6344.
[44] Gao Y,Zhao Y.Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing[J].J Integr Plant Biol,2013,56(4):343-349.
[45] Pattanayak V,Lin S,Guilinger JP,et al.High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity[J].Nat Biotechnol,2013,31(9):839-843.
[46] Lin Y,Cradick TJ,Brown MT,et al.CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences[J].Nucleic Acids Res,2014,42(11):7473-7485.
[47] Klug A.The discovery of zinc fingers and their applications in gene regulation and genome manipulation[J].Annu Rev Biochem,2010,79:213-231.
[48] Li T,Huang S,Zhao X,et al.Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes[J].Nucleic Acids Res,2011,39(14):6315-6325.
[49] Xu K,Ren C,Liu Z,et al.Efficient genome engineering in eukaryotes using Cas9 fromStreptococcusthermophilus[J].Cell Mol Life Sci,2015,72(2):383-399.
[50] Fu Y,Sander JD,Reyon D,et al.Improving CRISPR-Cas nuclease specificity using truncated guide RNAs[J].Nat Biotechnol,2014,32(3):279-284.
[51] Cho SW,Kim S,Kim Y,et al.Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases[J].Genome Res,2014,24(1):132-141.
[52] Ran FA,Hsu P,Lin CY,et al.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome gditing specificity[J].Cell,2013,154(6):1380-1389.
猜你喜欢
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
山东冶金(2018年5期)2018-11-22 05:12:46
西安建筑科技大学学报(自然科学版)(2016年1期)2016-11-08 12:15:18
中华灾害救援医学(2015年7期)2016-01-07 05:45:21
铁道科学与工程学报(2015年4期)2015-12-24 12:11:01
医学研究杂志(2015年7期)2015-06-22 11:01:01
现代检验医学杂志(2015年2期)2015-02-06 02:00:56
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
