
UFLC法同时测定甘草及其炮制品中2种活性成分的含量
2018-06-29 02:55:06孙宇慧王黎明郝乘仪朱鹤云
吉林医药学院学报 2018年4期
关 皎,王 强,孙宇慧,费 雪,王黎明,郝乘仪,冯 波,朱鹤云
(吉林医药学院药学院,吉林 吉林 132013)
甘草在中药传统方剂中频繁使用,有“十方九草”之说。甘草具有补脾益气、淸热解毒、祛痰止咳、缓急止痛、调和诸药的功效[1]。甘草中主要含有三萜皂苷类、黄酮类、生物碱类和多糖类成分,其中三萜皂苷类(代表药物为甘草酸)和黄酮类(代表药物为甘草苷)为甘草中的主要活性成分。研究表明,甘草酸具有抗病毒、抗炎、抗肿瘤、抗氧化、抗凝血、保肝、调节免疫、促进吸收等多种药理活性[2-3],甘草苷具有抗抑郁、保肝、神经保护等药理活性,同时对心肌缺血具有较好的治疗作用[4-6]。目前关于甘草的质量研究已有多篇文献报道,采用的方法包括紫外法[7]、荧光法[8]、高效液相色谱法[9-10]等。然而,这些方法存在样品前处理方法复杂、方法专属性和灵敏度差、分析时间过长等缺点。超快速液相色谱(ultra-fast liquid chromatography,UFLC)技术由于存在分析效率高、平衡时间短、溶剂消耗少等优点,近年来广泛应用于中药及中药复方的化学分析研究[11-12]。本研究首次采用UFLC法同时测定甘草及其炮制品中甘草苷和甘草酸的含量,旨在为甘草的药效物质基础和质量控制研究提供科学依据。
1 仪器与试药
日本岛津LC-20AD UFLC色谱仪系统,包括自动进样装置、二元高压输液泵、柱温箱,紫外检测器,LC solution色谱工作站;KQ-250DE型数控声波清洗器(昆山市超声仪器有限公司);CPA 225D型十万分之一电子天平(德国赛多利斯仪器有限公司)。
甘草药材(产地:内蒙古)购自吉林大药房,由吉林医药学院药剂学教研室李景华副教授进行鉴定,确定为豆科植物甘草GlycyrrhizauralensisFisch.的干燥根和根茎。炙甘草(产地:内蒙古)购自吉林大药房。
色谱纯乙腈和甲醇均购自美国Fisher公司,所用水为二次蒸馏水,其他试剂均为分析纯。对照品甘草苷(批号111610-201607)购自中国食品药品检定研究院,甘草酸(批号151014)购自成都普菲德生物技术有限公司,各对照品质量分数均大于98%。
2 方法与结果
2.1 对照品溶液的配制
分别精密称取甘草苷和甘草酸对照品适量,置于5 mL容量瓶中,用甲醇稀释至刻度,分别配制含甘草苷1.0 g/mL和甘草酸1.0 g/mL的对照品溶液备用。
2.2 供试品溶液的制备
称取预先通过40目筛的甘草药材粉末约0.2 g,置25 mL具塞碘量瓶中,加入50%乙醇25 mL,称定重量,超声提取30 min后,放冷,再次称定重量,并用50%乙醇补足减失的重量,摇匀,上清液用0.22 μm微孔滤膜滤过,取续滤液作为供试品溶液。
2.3 色谱条件
色谱柱为Shimadzu Shim-Pack XR-ODS柱(75 mm×3.0 mm,2.2 m);流动相为乙腈-0.05%磷酸水溶液,梯度洗脱:0~4 min,5%~20%乙腈;4~8 min,20%~40%乙腈;8~12 min,40%~45%乙腈,平衡时间2 min;体积流量0.8 mL/min;检测波长237 nm;柱温30 ℃;进样量5 μL。色谱图见图1。
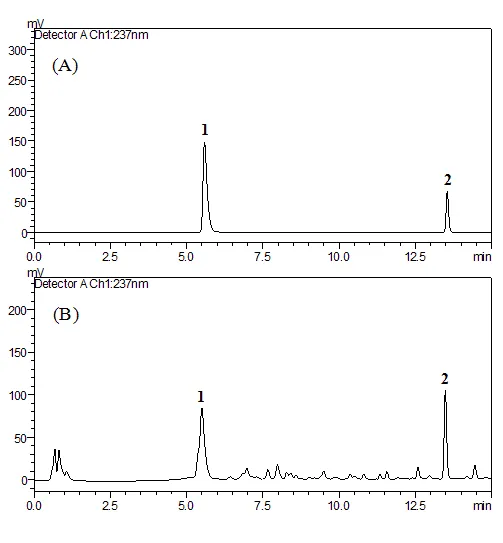
图 1 混合对照品(A)和甘草样品(B)的色谱图
2.4 线性关系考察
分别精密吸取“2.1”项下的制备甘草苷和甘草酸对照品溶液适量,置于相同5 mL容量瓶中,用甲醇-水(50∶50,v/v)配制成甘草苷和甘草酸浓度均为10、20、50、100、200、500 mg/L的混合对照品溶液,依次注入UFLC,以峰面积(Y)为纵坐标,对照品浓度X(mg/L)为横坐标,获得甘草苷和甘草酸的线性回归方程分别为:
Y=13 566X+59 140,R=0.999 8
Y=3 615.7X+10 400,R=0.999 9
结果表明,甘草苷与甘草酸浓度在10~500 mg/L范围内线性关系良好。
2.5 精密度实验
分别精密吸取混合对照品溶液(甘草苷与甘草酸浓度分别为100 mg/L)5 μL,连续进样6次,记录峰面积。结果甘草苷和甘草酸峰面积的RSD(n=6)分别为0.4%和0.4%,表明仪器精密度良好。
2.6 稳定性实验
取同一批次的甘草样品溶液,分别在0、2、4、8、12、24 h进样分析,记录峰面积。结果甘草苷和甘草酸峰面积的RSD分别为0.7%和1.9%,表明样品溶液在24 h内稳定。
2.7 重复性实验
取同一批甘草药材样品共6份,精密称定,分别制备样品溶液,进样分析,计算甘草苷和甘草酸的含量分别为1.85%和4.85%,RSD分别为1.6%和2.3%,表明方法的重复性良好。
2.8 加样回收率实验
精密称取已知含量(甘草苷和甘草酸含量分别为1.85%和4.85%)的甘草样品粉末9份,每份0.1 g,精密称定,分别加入相当于样品中甘草苷和甘草酸含量的80%、100%、120%的对照品溶液适量,各3份。制备样品溶液,进样分析,计算甘草苷和甘草酸的加样回收率和RSD,结果甘草苷的加样回收率为98.9%、99.5%、98.4%,RSD为1.5%、2.1%、0.9%;甘草酸的加样回收率99.2%、99.4%、99.4%,RSD为2.2%、1.3%、1.2%。
2.9 样品含量测定
精密称取6批甘草样品粉末(其中1~3批为生甘草,4~6批为炙甘草),制备样品溶液,进样分析,每个溶液进样3次,计算甘草苷和甘草酸的含量。样品测定结果见表1。
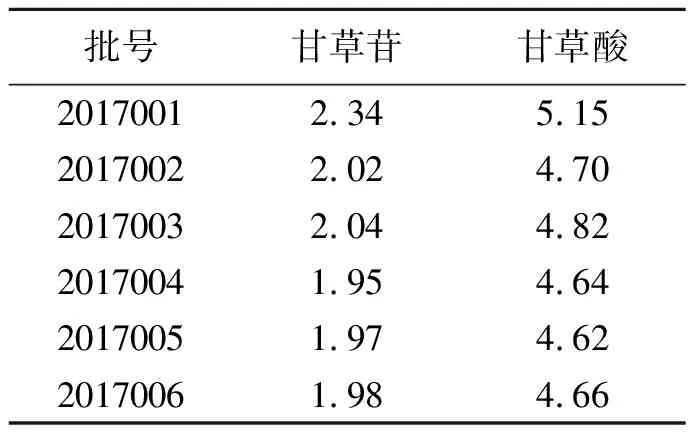
表 1 样品中甘草苷、甘草酸的含量测定结果(n=6,%)
3 讨 论
3.1 流动相的优化
实验比较考察了乙腈-水、甲醇-水、乙腈-1.0%甲酸水、乙腈-1.0%乙酸水、乙腈-0.05%磷酸水共5种流动相体系,结果表明,在确定的波长下,乙腈-0.05%磷酸水所得基线更平稳,各色谱峰的分离度更好,且色谱分析时间仅为15 min。
3.2 检测波长的选择
采用PDA检测器对样品在200~400 nm波长范围内进行扫描,甘草苷和甘草酸分别在276 nm和248 nm处有最大吸收。综合比较发现237 nm波长下检测的色谱图基线平稳,各主要特征峰吸收强度较大,故选定该波长作为检测波长。
3.3 提取方法及提取溶剂的选择
实验中考察了回流法和超声法提取甘草中甘草苷和甘草酸的提取效果,采用超声提取法时两种活性成分的含量明显高于回流提取法。本实验对比不同比例乙醇溶液和不同比例甲醇溶液对甘草中2种活性成分的提取效率,结果表明50%乙醇的综合提取效率最高,故本实验采用50%乙醇作为提取溶剂。
本研究采用UFLC法同时测定了3批生甘草和3批炙甘草中上述2种活性成分的含量。购自吉林大药房的3批生甘草和3批炙甘草的含量均符合药典要求(药典规定甘草中甘草苷含量不低于0.50%,甘草酸含量不低于2.0%;炙甘草中甘草苷含量不低于0.50%,甘草酸含量不低于1.0%)。但炮制后甘草中2种活性成分含量下降,与文献报道结果一致[11],甘草炮制后含蜂蜜固形物量增加有关约为11.8%,若扣除含蜂蜜固形物的量,炮制后甘草苷和甘草酸的平均含量均高于生品,由此可见甘草经蜜炙后入药具有一定的科学性。
参考文献:
[1] 国家药典委员会.中华人民共和国药典:2015年版一部[S].北京:中国医药科技出版社,2015:86-88.
[2] 史晓月.甘草酸药理机制的研究进展[J].中外医疗,2010,29(16):125.
[3] 张明发,沈雅琴.甘草酸抗病毒药理研究进展[J].中国执业药师,2008,5(12):18-22.
[4] 肖渊.甘草苷的抗抑郁作用及其机理研究[D].北京:北京中医药大学,2009.
[5] 丁选胜,戴德哉.甘草苷对CCl4肝脏毒性的保护作用[J].中药药理与临床,2002,18(6):12-13.
[6] 苏国林,刘刚,刘育辰,等.甘草苷的提取纯化方法和药理作用研究进展[J].中国现代中药,2013,13(10):48-51.
[7] 冯薇,王文全,赵平然.甘草总黄酮含量测定方法研究[J].时珍国医国药,2007,18(11):2608-2610.
[8] 孙艳涛,赵月,任晓宇,等.荧光光谱法测定甘草中甘草苷的含量[J].医药导报,2013,32(9):1210-1213.
[9] 刘雅茜,王梦月,史海明,等.高效液相色谱法测定甘草及其炮制品中5种活性成分的含量[J].中国药学杂志,2008,43(16):1268-1271.
[10] 章厉劼,孙真峥.HPLC法测定四君子散中甘草酸的含量[J].黑龙江畜牧兽医,2017,59(7):286-288.
[11] 李国俊.高效液相色谱法快速检测中药复方炮制液中芍药苷含量[J].河北省科学院学报,2013,30(3):59-62.
[12] 李启艳,王荣梅.快速溶剂萃取-超高效液相色谱法测定稳心颗粒中皂苷类成分[J].药物分析杂志,2015,35(3):528-531.
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
基层中医药(2021年3期)2021-11-22 08:08:04
中老年保健(2021年9期)2021-08-24 03:51:00
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
西南医科大学学报(2016年4期)2016-01-03 01:26:29
中国当代医药(2015年33期)2015-03-01 02:09:17
中国药业(2014年20期)2014-05-17 03:13:44
中医研究(2014年2期)2014-03-11 20:28:19
中国现代中药(2012年6期)2012-10-30 01:38:18
