
常压高浓度氧对脑缺血-再灌注大鼠缺血核心区核转录因子-κB的适度调节作用
2018-06-23 08:58师文娟赵咏梅戚智锋黄语悠房亚兰刘克建
首都医科大学学报 2018年3期
师文娟 赵咏梅 戚智锋 黄语悠 房亚兰 刘克建
(首都医科大学宣武医院 北京市老年病医疗研究中心 脑血管病转化医学北京市重点实验室,北京100053)
缺血性脑卒中是一种由于脑组织局部区域血液供应障碍,导致脑组织缺血缺氧性病变坏死、进而产生神经功能缺失的高发疾病。缺血再灌注过程中涉及炎性级联反应,导致神经细胞死亡或凋亡,加重缺血性损伤,因此干预脑缺血后的炎性反应是较为有效的治疗方案。脑缺血炎性反应受到多种基因调节,其中核转录因子-κB(nuclear factor-κB, NF-κB)作为细胞内信号转导的枢纽,在炎性反应中起关键作用,成为抗脑缺血炎性损伤的重要靶点[1]。
在神经系统中,NF-κB 广泛分布于神经元、神经胶质细胞和血管内皮细胞中,由p65或p50亚基形成二聚体,最常见的NF-κB亚基组成形式为p65/p50或p65/p65[2]。脑缺血再灌注损伤时 NF-κB活化后转位进入细胞核并与特定的DNA序列结合,诱导启动靶基因的转录及表达,从而激活炎性反应因子及细胞因子,导致缺血后炎性级联反应的发生[3]。NF-κB主要受其抑制剂(inhibitor of κB,IκB)蛋白调节[4]。白细胞介素-1β(interleukin-1β, IL-1β)是一种可由NF-κB介导产生的重要炎性因子,它可通过促使白细胞浸润和血小板黏附内皮细胞以及其他多种途径造成细胞和组织的损害[5]。肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)也是重要的炎性反应介质,在缺血后受NF-κB调控可引起神经细胞的炎性损伤[6]。
常压高浓度氧(normobaric hyperoxia, NBO)治疗是一种能够直接改善缺血脑组织氧合状态的有效易行的疗法,在恢复脑组织氧分压方面有显著功效,这种保护归因于NBO 具有提高缺血脑组织能量代谢的能力,已经证实NBO治疗能增加大脑血流量和氧输送,从而可对脑缺血再灌注损伤涉及的一系列复杂病理过程予以干预保护[7]。然而方便易行的NBO能否成为抵抗缺血炎性反应的潜在治疗方式从而用于脑卒中的救治值得探究,目前尚没有直接证据显示,NBO对NF-κB引发的缺血后炎性反应是否具有调节作用,本研究旨在利用大鼠脑缺血再灌注模型,研究NBO治疗对脑缺血核心区IL-1β因子、TNF-α因子、NF-κB蛋白及其抑制蛋白IκB的影响,为缺血性脑卒中治疗的干预策略提供新思路。
1 材料与方法
1.1 主要仪器设备
小动物麻醉机(Harvard Apparatus公司,美国)、手术显微镜(Carl Zeiss公司,德国)、双极电凝器(德威公司,中国)、冷冻切片机(Thermo Fisher Scientific公司,美国)、正置荧光显微镜(Nikon公司80i,日本)、化学发光系统(Chemi Scope公司,中国)。
1.2 实验材料
SPF级雄性SD大鼠15只,体质量280~320 g,由北京维通利华实验动物技术有限公司提供,实验动物许可证号:SCXK(京)2014-0001。恩氟烷购自河北一品制药公司;NF-κB p65抗体、IκB抗体、IL-1β抗体、TNF-α抗体购自美国CST公司;OCT冰冻切片包埋剂、GAPDH抗体、HRP标记山羊抗小鼠/ 兔二抗购自北京中杉金桥公司;抗兔荧光二抗购自美国Invitrogen公司等。
1.3 动物模型制作及取材
依据数字表法随机将SD大鼠分为Sham组(n=3)、Normoxia组(n=6)和NBO组(n=6)。称体质量后,先使用5%(体积分数)恩氟烷诱导麻醉大鼠,后使用2%(体积分数)恩氟烷混合70%(体积分数)N2O及30% (体积分数)O2维持麻醉状态。按照改良的Longa线栓法[8]制备大鼠大脑中动脉阻塞模型(middle cerebral artery occlusion, MCAO)缺血再灌注模型:于大鼠颈部正中切口,钝性分离暴露右侧颈总、颈内及颈外动脉;电凝颈外动脉后,在颈外动脉残端剪口,将线栓插入颈内动脉,至线栓顶端距离颈总动脉分叉处约1.8~2.0 cm且遇阻力感时停止。术中监测大鼠心率、血压及肛温,保持各项生理参数在正常范围。阻断血流90 min后,拔出线栓恢复血流灌注。Sham组大鼠进行手术及插栓操作后即刻拔出线栓恢复血供。NBO组大鼠于缺血5 min后,置于通入100%(体积分数)O2的麻醉盒中,其他两组大鼠置于空气环境。再灌注24 h处死大鼠取脑,于视交叉处作冠状切片,向前囟方向切2 片,小脑方向切4 片,每片厚度约2 mm。留取第3片脑组织用作Western blotting检测,第4 片脑组织速冻包埋后用作冰冻切片。依据文献[9-10]报道缺血核心区的病理特征及位置分布,选取缺血侧脑片中线旁开5.5 mm的外侧皮质为核心区范围。
1.4 Western blotting检测
选取缺血核心区脑组织,加入蛋白裂解液和蛋白酶抑制剂后置于冰上匀浆,冰浴30 min后于4 ℃,12 000 g离心30 min,BCA法测定上清液蛋白浓度。加入上样缓冲液,95 ℃水浴10 min,行SDS-PAGE电泳分离,以90 V恒定电压转膜100 min。5%(质量分数)脱脂牛奶室温封闭1 h,分别置于对应一抗溶液中4 ℃孵育过夜,NF-κB p65抗体(1∶1 000),IκB抗体(1∶1 000),GAPDH抗体(1∶1 000)。次日以TBST漂洗印迹膜5 min,重复3次;加入HRP标记的山羊抗小鼠/兔二抗(1∶6 000),室温孵育1 h。吸去二抗,以TBST漂膜5 min,重复3次;在膜上滴加化学发光液显影,使用化学发光系统扫描分析。蛋白水平用NF-κB p65或IκB与相应GAPDH的条带灰度比值表示。
1.5 免疫荧光检测
大鼠脑组织经OCT包埋后,行厚度为20 μm的冰冻切片。将切片置于预冷丙酮中固定10 min,PBS洗两次,加0.3%(体积分数)TritonX-100对切片进行破膜通透10 min,PBS洗两次,加5%(质量分数)BSA室温封闭1 h后分别滴加对应一抗 (NF-κB 1∶200, IL-1β 1∶200,TNF-α 1∶200),4 ℃ 孵育过夜。 PBS漂洗切片3次,每次5 min;滴加荧光二抗(1∶300),室温避光孵育1 h;PBS漂洗3次,每次5 min;用含DAPI染料的防荧光淬灭封片剂封片,在荧光显微镜下观察拍照。
1.6 统计学方法
2 结果
2.1 NBO治疗抑制大鼠脑缺血核心区IL-1β的表达
免疫荧光染色结果显示,Sham组大鼠大脑组织偶见微弱荧光染色。与Sham组相比,Normoxia组大鼠缺血核心区IL-1β阳性细胞数目增多,差异有统计学意义(P<0.05);与Normoxia组相比,NBO组大鼠缺血核心区IL-1β阳性细胞数目减少,差异有统计学意义(P<0.05),荧光强度也有所减弱(图1)。
2.2 NBO治疗抑制大鼠脑缺血核心区TNF-α的表达
免疫荧光染色结果显示,Sham组大鼠大脑组织偶见微弱荧光染色。Normoxia组大鼠缺血核心区TNF-α阳性细胞数目较Sham组增多,差异有统计学意义(P<0.05);NBO组大鼠缺血核心区TNF-α阳性细胞数目较Normoxia组减少,差异有统计学意义(P<0.05),荧光强度也有所减弱(图2)。
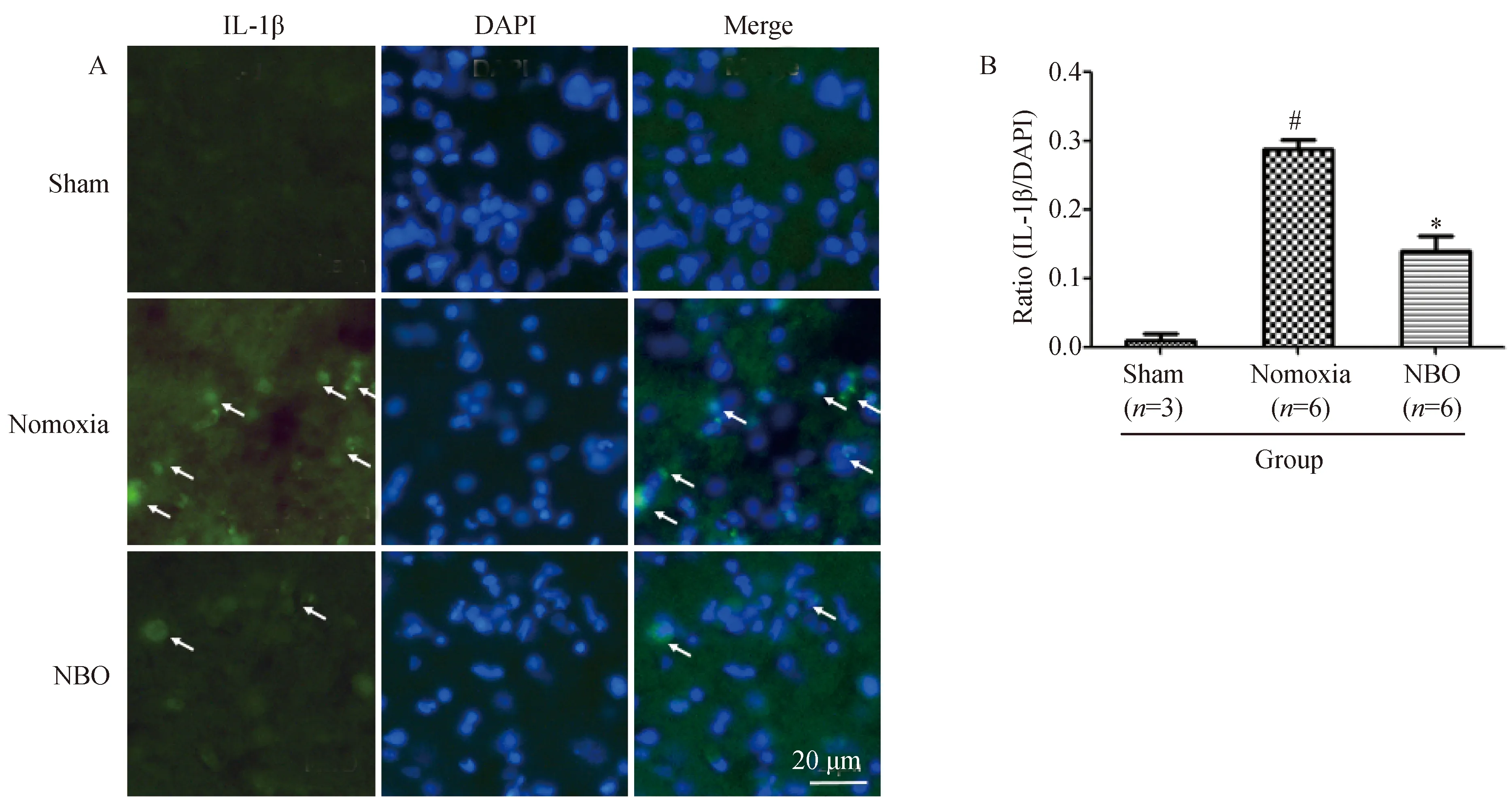
图1 免疫荧光检测大鼠脑缺血核心区IL-1β表达Fig.1 Immunofluorescence staining of IL-1β in the core ischemic region of rats following cerebral ischemia
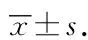
图2 免疫荧光检测检测大鼠脑缺血核心区TNF-α表达Fig.2 Immunofluorescence staining of TNF-α in the core ischemic region of rats following cerebral ischemia
2.3 NBO治疗对大鼠脑缺血核心区NF-κB活化有一定程度的抑制作用
免疫荧光结果显示,NF-κB p65被绿色荧光二抗标记,Sham组大鼠脑组织显示微弱荧光染色。不同实验组间NF-κB p65发生入核转移的阳性细胞数的差异代表其NF-κB活化程度有所不同。与Sham组相比,Normoxia组缺血核心区NF-κB p65荧光强度明显增强, NF-κB p65胞核染色阳性的细胞数量明显增多;与Normoxia组相比,NBO组缺血核心区NF-κB p65入核阳性细胞数目减少、荧光强度减弱,表明NF-κB活性受到一定程度的抑制,详见图3。
Western blotting检测结果显示与Sham组相比,实验组缺血核心区NF-κB p65浓度均有所升高;与Sham组相比,Normoxia组缺血核心区NF-κB p65蛋白浓度升高,差异有统计学意义(P<0.05);与Normoxia组相比,NBO组缺血核心区NF-κB p65蛋白浓度降低,差异有统计学意义(P<0.05),说明NBO对缺血-再灌注大鼠缺血NF-κB的活化有一定程度的抑制作用,详见图4A。
2.4 NBO治疗抑制大鼠脑缺血核心区IκB蛋白的降解
Western blotting检测结果显示:与Sham组相比,Normoxia组缺血核心区IκB蛋白浓度降低,差异有统计学意义(P<0.05);与Normoxia组相比,NBO组缺血核心区IκB蛋白浓度升高,差异有统计学意义(P<0.05);NBO组缺血核心区IκB蛋白浓度与Sham组差异不明显,详见图4B。
3 讨论
研究[11]表明炎性反应所致的继发性损害是脑缺血损伤的主要原因,由于核转录因子NF-κB可以调节多种炎性因子转录及表达,因而被认为是脑缺血后炎性级联反应的始动因素,参与了脑缺血损伤的发生及发展的主要过程,因此在脑缺血病程中对NF-κB进行干预可能会是较为有效的治疗策略。
NF-κB在神经元、 神经胶质细胞和脑血管内皮细胞中通常有较低量的表达,它在炎性反应因子的转录环节起着关键性作用。缺血时NF-κB被激活,活化的NF-κB可促使IL-1β、 TNF-α等炎性因子表达上调,反之这些因子也可以激活NF-κB,从而使NF-κB的活化在组织间不断扩散,加重炎性反应[12]。研究[13]表明抑制NF-κB活化是减轻脑缺血炎性损伤的重要途径,由于NF-κB是脑缺血炎性反应的中心环节,所以拮抗NF-κB的活化,可能起到有效的抗炎效果,从而发挥脑保护作用;而IκB的降解是NF-κB活化的重要条件,抑制或减少IκB降解可抑制NF-κB的活化[14]。本研究中NBO组缺血核心区IκB蛋白浓度较Normoxia组明显升高,说明NBO治疗显著抑制了缺血核心区IκB蛋白的降解。

图3 免疫荧光检测检测大鼠脑缺血核心区NF-κB p65活化水平Fig.3 Immunofluorescence staining of NF-κB p65 in the core ischemic region of rats following cerebral ischemia
然而,NF-κB在脑缺血病程中的作用具有双面性,一方面活化的NF-κB 通过促进炎性反应的发生发展、诱导细胞凋亡、介导自由基损伤等途径,加重脑缺血损伤[15];另一方面NF-κB的抑制细胞凋亡机制对于保护缺血性脑损伤具有重要作用[16]。因此适度抑制NF-κB 的活化对治疗脑缺血再灌注损伤疾病将会有很大价值,但同时需要关注的是还要将NF-κB的活性控制在一定的范围内,这将成为脑缺血性损伤疾病干预的新策略。本研究免疫荧光结果显示NBO治疗组缺血核心区NF-κB活化发生核转位的细胞数目较Normoxia组明显减少,但仍多于Sham组,说明NBO治疗既可维持NF-κB转录因子一定程度的激活状态,有利于抑制凋亡、启动机体自身保护机制;又可防止其过度活化,从而抑制炎性因子的表达,免疫荧光检测显示NBO治疗组缺血核心区IL-1β及TNF-α表达比Normoxia组显著减少,说明NBO治疗具有抗炎作用进而减轻脑缺血损伤。以上结果综合提示NBO治疗可能通过适度调节缺血核心区NF-κB的活化程度以发挥脑保护作用。
脑缺血性损伤根本致病原因是局部脑血管血流不畅导致的缺血缺氧,给予常压高浓度治疗恰是针对性地帮助缺血组织恢复氧分压,可以避免引入化学药物干预因素,减轻其他附加影响,可能对于一些在脑缺血再灌注损伤中具有双重作用的关键靶点起到适度调节的效果,有助于为脑缺血性损伤疾病干预方式提供新的思路和策略。
[1] Zhang S, Qi Y, Xu Y, et al. Protective effect of flavonoid-rich extract from rosa laevigata michx on cerebra ischemia-reperfusion injury through suppression of apoptosis and inflammation[J]. Neurochem Int,2013, 63(5): 522-532.
[2] Escalante C R, Shen L, Thanos D, et al. Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter[J]. Structure,2002,10(3):383-391.
[3] Zhan J, Qin W, Zhang Y, et al.Upregulation of neuronal zinc finger protein A20 expression is required for electroacupuncture to attenuate the cerebral inflammatory injury mediated by the nuclear factor-κB signaling pathway in cerebral ischemia/reperfusion rats[J]. J Neuroinflammation, 2016, 13(1): 258.
[4] Tsai C Y, Li F C, Wu C H, et al. Sumoylation of IκB attenuates NF-κB-induced nitrosative stress at rostral ventrolateral medulla and cardiovascular depression in experimental brain death[J]. J Biomed Sci, 2016,23(1):65.
[5] Cai Z, Zhang X, Wang G, et al. BDNF attenuates IL-1β-induced F-actin remodeling by inhibiting NF-κB signaling in hippocampal neurons[J]. Neuro Endocrinol Lett, 2014,35(1):13-19.
[6] Lykke K L, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke[J]. J Cereb Blood Flow Metab, 2012, 32(9): 1677-1698.
[7] Shi S, Qi Z, Ma Q, et al. Normobaric hyperoxia reduces blood occludin fragments in rats and patients with acute ischemic stroke[J]. Stroke, 2017,48(10):2848-2854.
[8] Tajiri N, Dailey T, Metcalf C, et al. In vivo animal stroke models: a rationale for rodent and non-human primate models[J]. Transl Stroke Res,2013, 4(3):308-321.
[9] 孙明,赵育梅,徐超. 局灶性脑缺血再灌注损伤核心区及半暗带的组织学定位[J].中华神经外科杂志,2003, 19 (4) :259-262.
[10] Dong W, Qi Z, Liang J, et al. Reduction of zinc accumulation in mitochondria contributes to decreased cerebral ischemic injury by normobaric hyperoxia treatment in an experimental strokemodel[J]. Exp Neurol, 2015, 272:181-189.
[11] Zhao Y, Fang Y, Li J, et al. Neuroprotective effects of chrysophanol against inflammation in middle cerebral artery occlusionmice[J]. Neurosci Lett,2016,630:16-22.
[12] Dadsetan S, Balzano T, Forteza J, et al. Reducing peripheral inflammation with infliximab reduces neuroinflammation and improves cognition in rats with hepatic encephalopathy. [J]. Front Mol Neurosci,2016,9:106.
[13] Liu W, Wang X, Zheng Y, et al. Electroacupuncture inhibits inflammatory injury by targeting the miR-9-mediated NF-κB signaling pathway following ischemicstroke[J]. Mol Med Rep,2016,13(2):1618-1626.
[14] Desai A, Singh N, Raghubir R. Neuroprotective potential of the NF-kappaB inhibitor peptide IKK -NBD incerebral ischemia -reperfusion injury[J]. Neurochem Int, 2010, 57 ( 8) : 876 -883.
[15] Zhang X, Zhang X,Wang C, et al. Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: Up -regulated pAkt, pGSK and pCREB, down-regulated NF-κB expression, ameliorated BBB permeability[J]. Brain Res, 2012, 1459: 61 -70.
[16] Ridder D A, Schwaninger M. NF-kappaB signaling in cerebral ischemia[J]. Neuroscience, 2009,158(3):995-1006.
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
昆明医科大学学报(2020年11期)2020-12-28
中成药(2018年4期)2018-04-26
种业导刊(2017年5期)2017-06-27
种业导刊(2017年4期)2017-06-19
种业导刊(2017年3期)2017-06-19
种业导刊(2016年9期)2016-01-03
中国康复理论与实践(2015年10期)2015-12-24
中国体外循环杂志(2015年3期)2015-12-08
医学研究杂志(2015年9期)2015-07-01
