
襄麦冬总甾体皂苷提取工艺优化及体外清除自由基活性评价
2018-06-19 09:50杨佳娣叶国栋潘婉莲余海忠
食品与机械 2018年4期
杨佳娣 柯 婉 叶国栋 潘婉莲 于 博 余海忠
(湖北文理学院化学工程与食品科学学院,湖北 襄阳 441053)
襄麦冬学名湖北麦冬(Liriopespicatavar.prolifera),为百合科山麦冬属植物,其块根与川麦冬、杭麦冬一起入药,连续收载于《中华人民共和国药典》(1995,2000,2005,2010,2015年版),是国家地理标志产品。甾体皂苷是襄麦冬的主要活性成分之一,在医学研究、保健品开发、食品生产等方面应用广泛[1]。目前,对襄麦冬甾体皂苷的研究,主要集中在成分或含量测定方面。张炜等[2]建立了UPLC-MS/MS法检测妇康宁片中掺加的湖北麦冬皂苷;吴发明等[3]从中国四大产地的麦冬中均检出麦冬皂苷D,但浙江麦冬含量较低;同时也发现不同产地麦冬中4种有效组分(2个皂苷和2个黄酮)的含量差异极显著[4]。陈乃东等[5]证明高效毛细管电泳法可用于25R与25S-鲁斯可皂苷元手性拆分,为鲁斯可皂苷元药理学研究、质量控制提供了新方法。吴弢等[6]采用ELSD测定了湖北麦冬中山麦冬皂苷 B的含量,结果表明ELSD是皂苷类化合物较为适宜的检测器。林以宁等[7]采用HPLC-ELSD法测定发现不同种麦冬类药材在皂苷元组成及含量上均有区别,其总苷元含量为:短葶山麦冬>川麦冬和湖北麦冬>杭麦冬。
也有学者开展了一些皂苷与其品质相关性的研究。王小刚[8]发现山麦冬皂苷B的含量在不同种苗间无显著差异,但是与土壤中有效磷和全钾呈显著性相关。兰宏昌[9]以山麦冬皂苷B含量等5个因素为考核指标,分析不同施加时间、不同施加剂量的多效唑对湖北麦冬质量和产量的影响,得出多效唑的合适施用量为0.84 g/m2,最佳施用时间为9月中下旬。朱业芹[10]以山麦冬皂苷B和多糖的含量作为质量评价指标,确定了以具有粗根、新生芽的植株为种苗,株距×行距(10 cm×15 cm)种植,摘花葶处理时的湖北麦冬质量和产量较优。
此外,在甾体皂苷的细胞活性评价、降血糖方面也有一些报道。林以宁等[7]发现不同种麦冬类药材均对中性粒细胞呼吸爆发呈抑制作用,其中,短葶山麦冬对中性粒细胞的呼吸爆发具有较强的抑制作用。刘霞等[11-12]证实了湖北麦冬所含总多糖和总皂苷均具有降血糖作用,降糖效果为湖北麦冬总多糖>湖北麦冬总皂苷>湖北麦冬总提取物。
目前,关于襄麦冬的皂苷提取研究报道较少,仅见林韵涵[13]与刘霞[11,14]的报道。前者通过正交试验获得总皂苷最佳提取工艺的得率为18.67%;后者则利用大孔吸附树脂分离、纯化襄麦冬块根提取物,得到纯度含量为66.52%的总多糖和81.15%的总皂苷。而对襄麦冬甾体皂苷的抗氧化活性研究则更为鲜见,仅见其不同极性部位粗提物的[15],尚未见单一类组分——总甾体皂苷的体外抗氧化活性报道。同时,从植物中寻找天然抗氧化物质已逐渐成为食品行业关注的热点[16-17],作为药食兼用的襄麦冬,不仅具有较好的药理与保健作用,还被广泛用于食品开发,目前已有麦冬酒、麦冬茶、麦冬咀嚼片、麦冬酸奶等产品问世[18-19]。
基于上述,本试验拟通过响应面法优化襄麦冬总甾体皂苷的提取工艺,并采用α-脱氧核糖法、邻苯三酚自氧化法、DPPH法、ABTS法测试其体外抗氧化能力,以期能为天然抗氧化剂的开发提供一种新的思路。
1 材料与方法
1.1 材料与仪器
襄麦冬块根:襄麦冬原产地——湖北襄阳欧庙,新鲜块根洗净后60 ℃烘干至恒重,粉碎待用;
α-脱氧核糖、硫代巴比妥酸、邻苯三酚、ABTS、生育酚、甲醇等:分析纯,国药集团上海化学试剂有限公司;
DPPH:分析纯,美国Sigma公司;
山麦冬皂苷B标准品:分析纯,中国食品药品检定研究院;
微型植物粉碎机:FZ102型,天津泰斯特仪器有限公司;
数显鼓风干燥箱:GZX-9240 MBE型,上海博讯实业有限公司医疗设备厂;
紫外-可见光分光光度计:UV-1800型,日本岛津公司;
高效液相色谱仪:LC-20AT型,日本岛津公司;
超声波清洗器:KH3200DB型,昆山禾创超声仪器有限公司;
小型冷冻干燥机:FreeZone 12型,美国Labconco公司。
1.2 襄麦冬总甾体皂苷的提取
分别称取1.0 g襄麦冬块根粉碎物,加入一定体积甲醇进行超声处理,在设定的温度下提取一定时间后,过滤。滤渣转移至索氏提取器内,加入100 mL甲醇,80 ℃恒温水浴,继续提取一定时间后停止,合并2次滤液,减压浓缩得浸膏。取100 mL纯水常温超声溶解浸膏,过滤,滤液中加入40 mL乙醚进行萃取脱酯。对保留的水层部分,依次加入15,10,5,5 mL水饱和正丁醇,分4次萃取样品溶液,合并萃取的正丁醇层部分,依次进行45 ℃水浴减压浓缩和-40 ℃真空冷冻干燥,所得产物即为襄麦冬总甾体皂苷。
1.3 甾体皂苷的HPLC含量测定
以山麦冬皂苷B的含量来示踪襄麦冬块根提取物中的总甾体皂苷含量,HPLC检测条件:检测器SPD-M20A,色谱柱Inert Sustain C18,柱温箱CTO-20A,真空脱气机DGU-20A3,流动相85%甲醇,流速0.3 mL/min,检测波长208 nm,进样量10 μL,等度洗脱40 min。用甲醇将山麦冬皂苷B标准品配制成0.125,0.250,0.050,0.750,1.000 mg/mL 的溶液,上机检测后以质量浓度为横坐标、峰面积为纵坐标制作标准曲线(图1),得到回归方程y=7.99×106x-1.08×106,R2=0.995。
1.4 襄麦冬总甾体皂苷的提取单因素试验设计
1.4.1 液料比对得率的影响 各取1 g样品,分别按液料比10∶1,15∶1,20∶1,25∶1,30∶1,35∶1 (mL/g)与甲醇混合,30 ℃超声处理1.0 h后转入索氏提取1.0 h。最后对样品进行萃取、浓缩、干燥,用30 mL甲醇溶解定容即得总甾体皂苷溶液,测定其含量。
1.4.2 超声处理温度对得率的影响 各取1 g样品,按液料比20∶1 (mL/g)与甲醇混合,分别在25,30,35,40,45,50 ℃下超声处理1.0 h后转入索氏提取1.0 h。最后对样品进行萃取、浓缩、干燥,用30 mL甲醇溶解定容即得总甾体皂苷溶液,测定其含量。
1.4.3 超声处理时间对得率的影响 各取1 g样品,按液料比20∶1 (mL/g)与甲醇混合,30 ℃分别超声处理0.5,1.0,2.0,3.0,4.0,5.0 h后转入索氏提取1.0 h。最后对样品进行萃取、浓缩、干燥,用30 mL甲醇溶解定容即得总甾体皂苷溶液,测定其含量。

图1 山麦冬皂苷B的HPLC色谱图及标准曲线
1.4.4 索氏提取时间对得率的影响 各取1 g样品,按液料比20∶1 (mL/g)与甲醇混合,30 ℃超声提取1.0 h后转入索氏提取0.5,1.0,2.0,3.0,4.0,5.0 h。最后对样品进行萃取、浓缩、干燥,用30 mL甲醇溶解定容即得总甾体皂苷溶液,测定其含量。
1.5 甾体皂苷的抗氧化活性测试

1.5.1 清除OH自由基的活性测试 采用α-脱氧核糖法[20]。取不同的具塞试管,分别依次加入0.20 mL的FeSO4-EDTA溶液(10 mmol/L)、0.20 mL的α-脱氧核糖溶液(10 mmol/L)、1 mL不同浓度的样品溶液、0.4 mL磷酸缓冲液(pH 7.4,0.1 mol/L)和0.20 mL H2O2(10 mmol/L),混匀,37 ℃水浴1 h后,再依次加入1 mL的2.8 g/100 mL三氯乙酸溶液和1.0 mL的1.0 g/100 mL硫代巴比妥酸溶液,迅速摇匀,100 ℃水浴10 min,冷水冷却后在532 nm处测定吸光值AS。不加样品溶液,同上操作处理,测定其对比吸光值AC。不加样品溶液且不在37 ℃水浴中反应,其它处理同上,测定空白吸光值A0。每个样品重复3次,清除率按式(1)计算:
S=[1-(AS-A0)/(AC-A0)]×100%,
(1)
式中:
S——清除率,%;
AS——样品吸光值;
A0——空白吸光值;
AC——对比吸光值。
S=(Ac-As)/Ac×100%,
(2)
式中:
S——清除率,%;
Ac——对照吸光值;
As——样品吸光值。
1.5.3 清除DPPH自由基的活性测试 采用DPPH法[22]。取不同的具塞试管,分别依次加入3.9 mL的DPPH甲醇溶液(25 mg/L)和0.1 mL不同浓度的样品,混匀,将试管放入暗处反应30 min后,立刻取出测定其在515 nm处吸光值,空白用甲醇替代样品溶液,其它操作相同。每个样品重复3次,清除率按式(3)计算:
S=[(A0-As)/A0]×100%,
(3)
式中:
S——清除率,%;
As——样品吸光值;
A0——空白吸光值。
1.5.4 清除ABTS自由基的活性测试 采用ABTS法[23]。取0.2 mL的ABTS(7.4 mmol/L)溶液,加入0.2 mL的K2S2O8(2.6 mmol/L)溶液,混匀,室温避光静置12 h,用95%乙醇稀释40倍,此时在734 nm处测得吸光值为0.7±0.02即可。取不同的具塞试管,分别加入0.8 mL上述溶液和0.2 mL 不同浓度的样品溶液,剧烈振摇10 s,充分混合,静置6 min,立刻测定其在734 nm处吸光值。空白用乙醇代替,每个样品重复3次,清除率按式(4)计算:
S=(1-AS/A0)×100%,
(4)
式中:
S——清除率,%;
AS——样品吸光值;
A0——空白吸光值。
1.6 数据处理
响应面试验设计与数据处理采用Design-expert 8.0.6进行,其它数据采用Origin Pro 8.5进行分析处理。
2 结果与分析
2.1 单因素试验结果
目前,已从襄麦冬块根中分离出包括山麦冬皂苷B在内的14种结构的甾体皂苷[24]。本试验以山麦冬皂苷B含量为示踪指标。
2.1.1 液料比对得率的影响 由图2(a)可知,液料比对甾体皂苷含量影响较大。总体上看,在设定的范围内液料比为20∶1 (mL/g)时,提取物中山麦冬皂苷B的含量最大,达到0.069 2 mg/mL。此时,也意味着提取的甾体皂苷的含量最大。
2.1.2 超声处理温度对得率的影响 由图2(b)可知,超声温度对山麦冬皂苷B的影响趋势与液料比的类似,也是先增加后减小,可能与襄麦冬块根中富含多糖有一定的关系,在加热的情况下,样品粉末自身携带的少量水分与富含多糖的块根粉末糊化,形成胶体,阻挡了部分甲醇与甾体皂苷分子的充分接触,影响了甾体皂苷的进一步溶解。
2.1.3 超声处理时间对得率的影响 由图2(c)可以看出,超声处理时间对甾体皂苷的提取含量的影响比较小,提取物中山麦冬皂苷B的平均含量维持在0.058 5 mg/mL左右,因此,取0.5 h作为超声处理时间。
2.1.4 索氏提取时间对得率的影响 由图2(d)可知,在3 h之前,随着提取时间的延长,山麦冬皂苷B的含量也随之提高,达到3 h之后,提取含量的增幅减小,甾体皂苷的含量趋于稳定。因此,考虑提取时间成本,选择3 h作为最佳索氏提取时间。
2.2 响应面法试验结果
2.2.1 响应面法试验设计及结果 利用Design-expert软件,结合单因素试验结果,在超声处理0.5 h的基础上,选择液料比、超声处理温度、索氏提取时间3个为影响因素,分析各因素及因素间对提取物中山麦冬皂苷B含量的影响,试验设计及结果分别见表1、2。

图2 处理因素对襄麦冬总甾体皂苷含量的影响
2.2.2 方差分析 利用软件对表2试验数据进行多元回归比拟,得出甾体皂苷含量(Y)对超声处理液料比、超声处理温度和索氏提取时间的二次多项回归方程为:
Y=-0.221 65+0.008 435A+0.008 79B+0.025 35C+3E-06AB-0.000 485AC-0.000 185BC-0.000 182 5A2-0.000 104 5B2-0.001 262 5C2。
(5)
由表3可以看出,模型P=0.000 3<0.01,说明此回归模型极显著;失拟误差P=0.797 1>0.05,说明检验结果与模型计算结果不存在显著差异,相关系数R2=0.964 2,说明96.42% 的试验数据的变异性可以用此回归模型解释,和实际情况拟合程度较好。因此,该方程可用于分析和预测襄麦冬块根总甾体皂苷的提取工艺。
2.2.3 主效应分析 对试验数据进行方差分析,结果见表3。根据各因素变量的显著性检验,可以看出:C因素的显著水平P<0.01,说明索氏提取时间对甾体皂苷含量的影响极显著;A因素的显著水平P<0.05,说明超声液料比对甾体皂苷含量的影响显著;而B因素显著水平0.193 2>0.05,即超声处理温度对甾体皂苷含量的影响不显著,因此,各因素对甾体皂苷含量的影响主次顺序为:C>A>B。

表1 Box-Behnken设计的因素水平表
表2 Box-Behnken的试验设计及结果
Table 2 Experimental design and results of Box-Behnken center composite
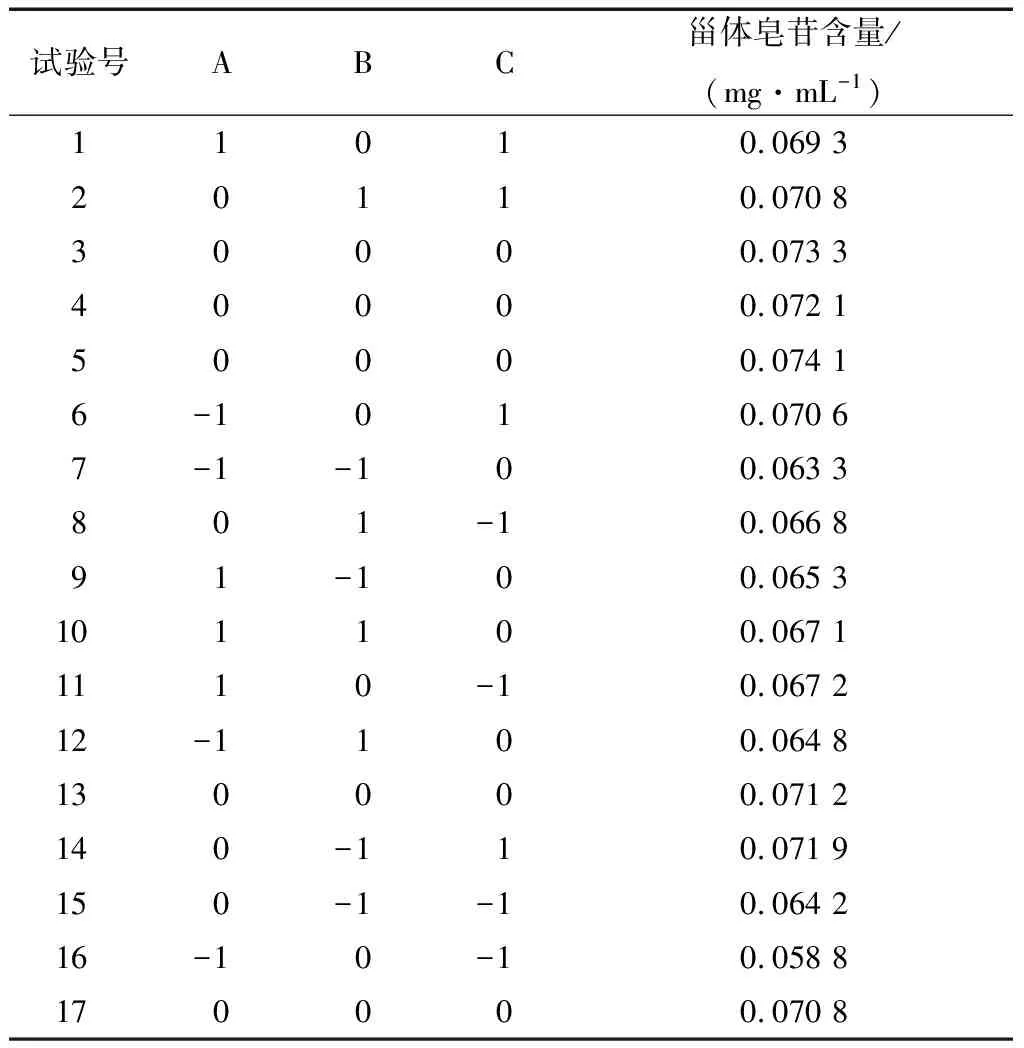
试验号ABC甾体皂苷含量/(mg·mL-1)11010.069 320110.070 830000.073 340000.072 150000.074 16-1010.070 67-1-100.063 3801-10.066 891-100.065 3101100.067 11110-10.067 212-1100.064 8130000.071 2140-110.071 9150-1-10.064 216-10-10.058 8170000.070 8
2.2.4 两因素间的交互效应分析 保持1个因素为最优条件,对其它2个因素间的交互效应进行分析,它们与响应值的关系用三维空间响应面图和三维等高线图表示,具体见图3。
图3(a)、(b)显示的是超声处理温度与液料比对甾体皂苷含量的影响。随着温度和液料比的增加,甾体皂苷含量均有一个先逐渐升高后逐渐降低的过程。图3(c)、(d)显示的是索氏提取时间与液料比对甾体皂苷含量的影响。当固定液料比在某一值时,随着索氏提取时间的延长,甾体皂苷含量逐渐升高;当固定索氏提取时间在某一值时,随着液料比的升高,甾体皂苷含量先增加后减少。图3(e)、(f)显示的是索氏提取时间与超声处理温度对甾体皂苷含量的影响,它们之间的交互影响趋势与图3(c)、(d)类似。
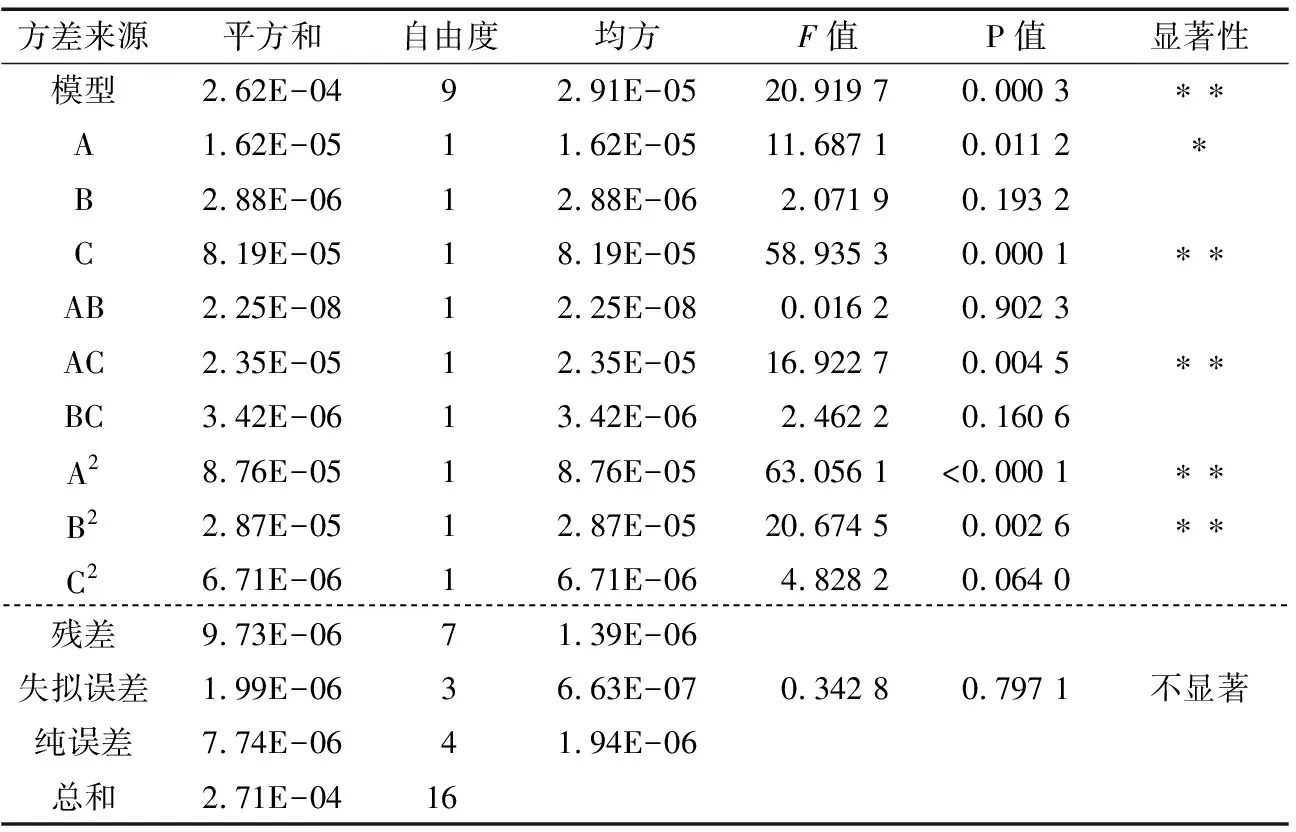
表3 各因素的回归系数及影响因子的显著性†
† *:P<0.05,显著;**:P<0.01,极显著;R2=0.964 2。
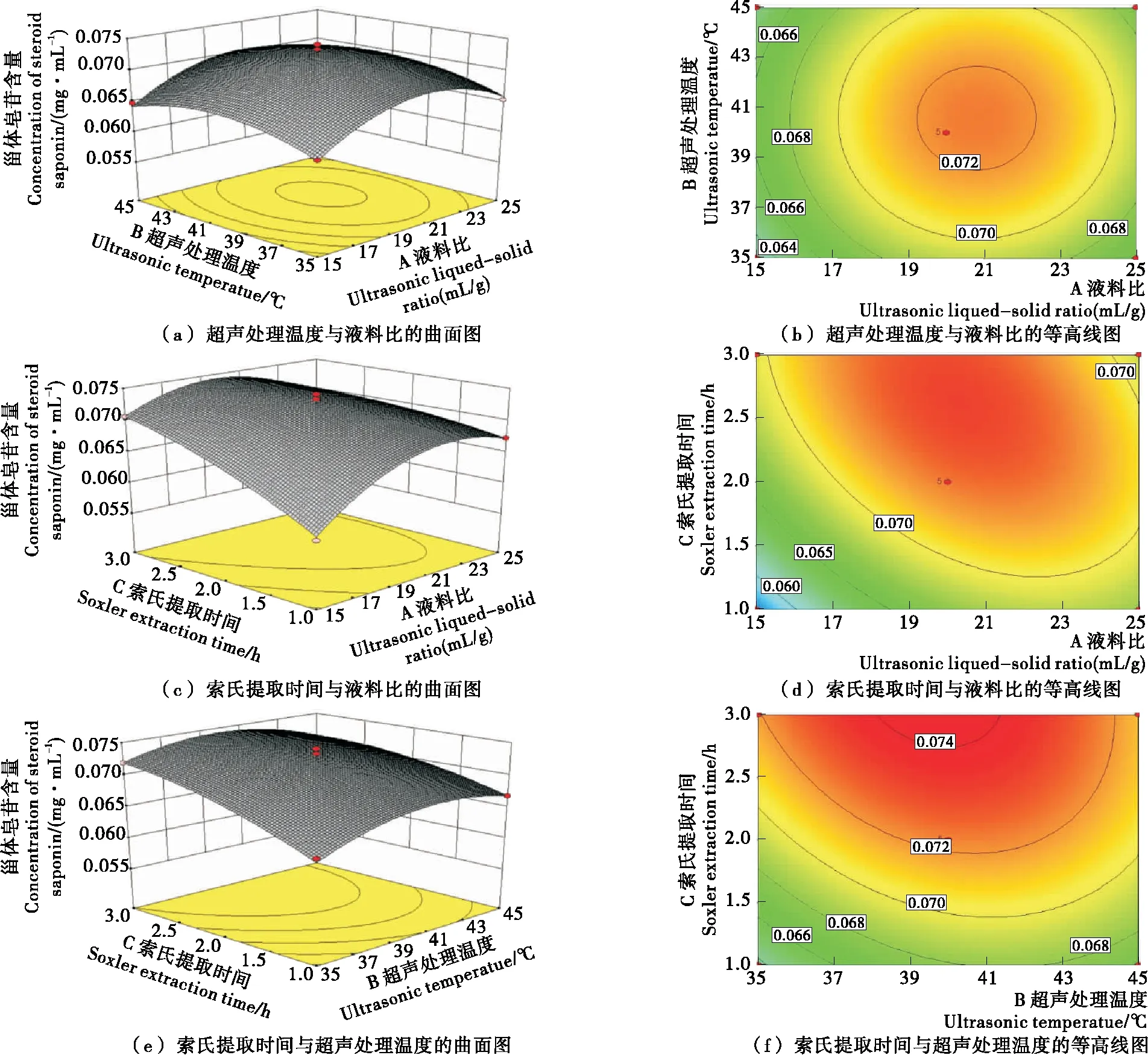
图3 两因素间的交互效应分析
2.2.5 响应面最佳条件的选择及验证 利用软件对襄麦冬总甾体皂苷提取工艺进行响应面分析,得出最佳提取条件为:超声处理时间0.5 h、液料比19.45∶1 (mL/g)、超声处理温度39.68 ℃、索氏提取时间3 h,此时,提取物中甾体皂苷含量最高,达到0.074 3 mg/mL。为了验证预测值是否准确,同时考虑到实际操作的便利,将最佳条件调整为:超声处理时间0.5 h、液料比19.5∶1 (mL/g)、超声处理温度39.7 ℃、索氏提取时间3 h。在此条件下,进行3次平行实验,结果测得的甾体皂苷含量为0.073 8 mg/mL,与预测值相差0.000 5,非常接近。这证明二次多元回归模型的预测值与实际值吻合良好,具有较高的可信度和实用价值。本试验主要考察响应面法在襄麦冬甾体皂苷提取条件上的优化应用,对样品采用甲醇溶解辅以超声处理、80 ℃索氏提取、萃取、减压浓缩和冷冻干燥等处理步骤,获得最佳工艺下的提取得率为22.14%,比林韵涵等[13]报道的通过正交试验优化的提取得率(18.67%)高出近4%,证明采用响应面法优化可有效提高襄麦冬甾体皂苷的得率。
2.3 自由基清除活性的评价
2.3.1 清除OH自由基的活性测试 图4(a)显示的是甾体皂苷对OH自由基的清除活性变化情况:在较低浓度范围内,清除率随着浓度的增加而急剧升高;但是当浓度达到一定数值后,清除率虽然仍随浓度的增加而升高,但是升高的趋势减缓,这种变化趋势与阳性对照生育酚类似。在设定的最高浓度为1.28 mg/mL时,清除率达到最高(59.66%),表明襄麦冬甾体皂苷对OH自由基具有较强的清除能力。
2.3.3 清除DPPH自由基的活性测试 图4(c)显示的是襄麦冬甾体皂苷对DPPH自由基的清除活性。虽然随着样品浓度的升高而呈逐步加强的趋势,但是总体上看,相较于阳性对照生育酚,其清除能力并不强,在设置的最高浓度1.28 mg/mL时,清除率也仅为10.5%。
2.3.4 清除ABTS自由基的活性测试 由图4(d)可知,同清除DPPH自由基活性一样,襄麦冬甾体皂苷对ABTS自由基的清除能力也较弱,在设置的最高浓度1.28 mg/mL时,清除率也仅为28.7%。

图4 襄麦冬甾体皂苷对不同自由基的体外清除活性评价
3 结论
本试验以湖北道地药材——襄麦冬块根为研究材料,采用响应面法优化了总甾体皂苷提取工艺,并对提取物的体外清除自由基能力进行了评价。结果表明,当采用超声处理时间0.5 h、液料比19.5∶1 (mL/g)、超声处理温度39.7 ℃、索氏提取时间3 h处理襄麦冬块根粉碎物时,提取物中甾体皂苷含量最高,此时,山麦冬皂苷B可达到0.073 8 mg/mL,即30 mL的提取物溶液中山麦冬皂苷B的含量为2.21 mg,提取得率达到22.14%。
本试验对襄麦冬甾体皂苷所做的提取工艺优化,仅仅是采用响应面法进行的前期实验室研究,考虑到工业生产应用实际,下一步的工作将从提取、分离、纯化融合工艺参数优化,以及降低生产成本等方面展开,以期能建立一套产能高、成本低的成熟生产工艺。
[1] 刘星, 余江丽, 刘敏, 等. 近10年甾体皂苷的生物活性研究进展[J]. 中国中药杂志, 2015, 40(13): 2 518-2 523.
[2] 张炜, 宋文静, 武嘉庚, 等. UPLC-MS/MS法检测妇康宁片中掺加的山麦冬[J]. 中成药, 2017, 39(4): 867-869.
[3] 吴发明, 杨瑞山, 李敏, 等. 麦冬主流品种药材质量比较研究[J]. 中国药学杂志, 2017, 52(6): 447-451.
[4] 吴发明, 张思荻, 曾俊, 等. HPLC-ELSD法测定不同产地麦冬中4种代表性成分的含量[J]. 药物分析杂志, 2016, 36(8): 1 370-1 376.
[5] 陈乃东, 陈琼, 何健. 高效毛细管电泳法拆分25R、25S鲁斯可皂苷元初步研究[J]. 药物分析杂志, 2014, 34(5): 805-812.
[6] 吴弢, 余伯阳, 程志红, 等. HPLC-ELSD法测定湖北麦冬中主要皂苷的含量[J]. 中草药, 2000, 31(3): 175-177.
[7] 林以宁, 朱丹妮, 寇俊萍, 等. 麦冬类药材皂苷元含量与其抑制中性粒细胞呼吸爆发的相关性[J]. 中国药科大学学报, 2007, 38(6): 549-552.
[8] 王小刚. 湖北麦冬种植优化和遗传多样性研究[D]. 武汉: 华中科技大学, 2011: 25-30.
[9] 兰宏昌. 多效唑应用于湖北麦冬种植的实验研究[D]. 武汉: 华中科技大学, 2010: 46-48.
[10] 朱业芹. 湖北麦冬关键种植技术研究[D]. 武汉: 湖北中医学院, 2007: 28, 35, 39.
[11] 刘霞. 湖北麦冬规范化种植及其提取物的降血糖作用研究[D]. 武汉: 湖北中医学院, 2009: 41-44.
[12] 刘霞, 张安萍, 孙江桥, 等. 湖北麦冬不同提取物降血糖作用的研究[J]. 中国医院药学杂志, 2009, 29(9): 719-721.
[13] 林韵涵, 李崇明, 李晓东, 等. 湖北麦冬有效部位总皂苷的提取纯化工艺研究[J]. 中药材, 2013, 36(5): 803-806.
[14] 刘霞, 张琼光, 向阳, 等. 大孔吸附树脂同步提取湖北麦冬总多糖和总皂苷[J]. 时珍国医国药, 2009, 20(5): 1 235-1 236.
[15] 聂伦, 熊尚森, 余海忠. 襄麦冬不同极性部位粗提物抗氧化性能初步评价[J]. 山地农业生物学报, 2013, 32(1): 32-34, 51.
[16] 葛智超, 陈宏伟, 朱蕴兰, 等. 富锗金针菇菌丝体多糖的分离纯化及对自由基的清除能力[J]. 农业工程, 2017, 7(3): 100-104, 112.
[17] 祁小妮, 马晓红, 李振亮, 等. 响应曲面优化丹参多糖提取工艺及抗氧化活性分析[J]. 南方农业学报, 2016, 47(11): 1 926-1 931.
[18] 邹光友. 麦冬须根食用的综合开发利用[J]. 食品科学, 1992(2): 33-36.
[19] 陈开霜, 陈美芳, 于博, 等. 转谷氨酰胺酶在凝固型麦冬酸奶中的应用研究[J]. 中国酿造, 2017, 36(9): 158-162.
[20] 周婷, 刘统, 林丹洁, 等. 鱼腥草甲醇提取物体外抗氧化性及抑菌活性评价[J]. 山地农业生物学报, 2012, 31(1): 87-90.
[21] 陈留勇, 孟宪军, 贾薇, 等. 黄桃水溶性多糖的抗肿瘤作用及清除自由基、提高免疫活性研究[J]. 食品科学, 2004, 25(2): 167-170.
[22] 余海忠, 刘统, 林丹洁, 等. 鄂西北产鱼腥草提取物体外抗氧化性的四种化学方法评价[J]. 中国野生植物资源, 2012, 31(1): 22-25.
[23] 白海娜, 王振宇, 刘瑞海, 等. 白藜芦醇与黑木耳多糖协同清除ABTS自由基活性的研究[J]. 现代食品科技, 2014, 30(3): 64-68.
[24] YU Bo-yang, QIU Sheng-xiang, ZAW Kyaw, et al. steroidal glycosides from the subterranean parts ofLiriopespicatavar.prolifera[J]. Phytochemistry, 1996, 43(1): 201-206.
猜你喜欢
海南热带海洋学院学报(2021年5期)2021-11-07
森林工程(2021年4期)2021-08-23
作文与考试·小学低年级版(2021年12期)2021-08-09
魅力中国(2020年8期)2020-12-07
海洋通报(2020年2期)2020-09-04
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年5期)2018-06-13
学术交流(2018年7期)2018-02-20
中成药(2017年4期)2017-05-17
速读·中旬(2016年9期)2017-05-09
