
链穿梭聚合的研究进展*
2018-06-13 07:52:04成振美傅智盛范志强
弹性体 2018年3期
成振美,傅智盛,范志强
(浙江大学 高分子科学与工程学系,浙江 杭州 301127)
自20世纪50年代,Ziegler[1]和Natta[2]在烯烃聚合催化剂取得开创性工作以来,聚烯烃的科学研究已过去了半个多世纪。尽管Ziegler-Natta催化体系已经取得了巨大的科研成果,但人们仍旧致力于追求采用廉价且来源丰富的单体来合成具有新型结构、性能更加卓越的聚合物[3-4]。20世纪80年代中期,均相茂金属催化剂的开发和应用取得了突破性进展[5],是合成聚烯烃材料的又一次重大的创新。茂金属催化剂因其只有一个活性中心的特性,能够得到相对分子质量及其分布、共聚单体含量和结晶结构可控的聚合物[6],并且能催化α-烯烃单体聚合生成立构规整度极高的等规或间规聚合物[7-9]。例如,1995年Coates等[10]使用双(2-苯基茚基)二氯化锆催化丙烯聚合,当改变聚合时的压力和反应温度时,可以改变催化剂的立体选择性,合成立构嵌段聚丙烯。但茂金属催化剂价格比较昂贵,且难以实现工业化。1995年,Brookhart等[11]6414[12]开发了用于烯烃聚合的后过渡金属催化剂。采用Brookhart型的α-二亚胺镍/钯催化剂可合成较高相对分子质量的聚乙烯,并且可以通过调节聚合时的压力和反应温度,调节聚合物的支化度,制备线形到高度支化的聚乙烯,从而实现对聚乙烯拓扑结构的微观调控[13]。然而,α-二亚胺镍/钯催化剂耐高温性能差,当聚合温度高于60 ℃时,催化剂会迅速分解,不再具备催化活性。后来人们分别通过对与N相连的苯环[14]和α-二亚胺的骨架结构[15]进行修饰来提高α-二亚胺镍/钯催化剂的耐热性,在保证较高催化活性的条件下,聚合温度可提高至100 ℃。2006年陶氏化学首次提出“链穿梭聚合”概念[16]714,引起国内外科研工作者对烯烃嵌段共聚物的极大兴趣。如果可以通过“链穿梭聚合”的方法得到烯烃嵌段共聚物,不仅能够大大简化生产工艺、降低生产成本,还能对聚合物的微观结构进行调整,更易得到目标产物。与现有的聚烯烃弹性体(POE)相比,烯烃嵌段共聚物有着很多独一无二的特性:弹性与耐高温性平衡的改进;具有较高的结晶温度,加工时能快速成型;无论室温还是高温下,都具有更好的弹性恢复和压缩形变;耐磨性得到提高。烯烃嵌段共聚物可以用作热塑性弹性体材料、增溶剂、药物载体、高抗冲塑料等。本文着重介绍了链穿梭聚合的基本原理和链穿梭聚合的研究进展,并且对链穿梭聚合的发展前景进行了展望。
1 链穿梭聚合机理
采用烯烃活性聚合催化剂,通过向体系中不断添加单体的方法来合成烯烃嵌段共聚物[17],用这种聚合方法可以精确控制聚合物的链结构、相对分子质量及其分布。然而,活性烯烃聚合有其固有的缺陷:一方面,活性聚合催化效率低,一个催化剂分子只能产生一条聚合物链,并且在活性聚合后期一般需要复杂的功能化、提纯和分离等步骤,这些都将导致烯烃共聚物商业使用的高成本;另一方面,活性聚合一般需要比较低的聚合温度,致使生成的聚合物过早地从体系中沉淀出来,导致聚合物因结晶度过低性能受到极大的影响,这都会限制烯烃活性聚合的工业化[18]。为了显著减少昂贵的过渡金属催化剂的消耗量,促使每个催化剂分子可以产生几条大分子链,配位链转移聚合(CCTP)进入人们的视野,成为科学家重点研究的又一热门课题[19-21]。CCTP催化体系由过渡金属催化剂和链转移剂组成,聚合过程中,在催化剂上生长的聚合物链和链转移剂上的烷基发生快速交换,链转移剂上的烷基与中心金属相连形成新活性中心,继续引发链增长,而原聚合物链转移到烷基金属化合物上,形成休眠的聚合物链,新生成的聚合物链与休眠的聚合物链不断发生快速的链交换反应,使聚合物链长平均化,实现相对分子质量及其分布的可控[22]。根据CCTP理论,链转移反应不再要求链转移剂上的烷烃完全被高分子链取代, 这为链穿梭聚合理论的提出提供了理论依据。
1.1 链穿梭聚合基本原理
陶氏化学公司提出的链穿梭聚合体系[23]包括两种单体(乙烯和辛烯)、两种共聚能力不一样的催化剂(Cat.1、Cat.2结构式见图1)以及一种链穿梭剂二乙基锌(CSA)。所谓的链穿梭聚合即增长的聚合物链在多个催化活性中心之间穿梭,每一个聚合物链至少在两个催化活性中心上增长。链穿梭剂是一种金属烷基复合物,能够促进上述的链转移反应。根据两种催化活性中心对乙烯和辛烯选择性不同,可生成不同结晶度的链段。其中Cat.1对乙烯具有较高的选择性,生成结晶度较高的链段,称为“硬段”;Cat.2具有较好的共聚性能,生成玻璃化转变温度较低(Tg<-40 ℃)的无定形材料,称为“软段”。以上两种“软”、“硬”不同的链段,在有效的链穿梭剂存在下,经过链转移在不同活性中心上增长,就可制备出烯烃嵌段共聚物,链穿梭聚合原理见图2。

图1 链穿梭聚合选用的Cat.1和Cat.2

图2 链穿梭聚合原理示意图
1.2 催化剂和链穿梭剂选择的基本条件
Arriola等[16]715在较高温度下(>120 ℃)对催化剂和链穿梭剂采用高通量筛选的方法,经过1 600多次的筛选工作,最终确定能够较好符合聚合条件及催化性能的两种催化剂(Cat.1和Cat.2)和CSA。由此可以看出,链穿梭聚合所采用催化剂和链穿梭剂的筛选条件是非常严苛的:(1)由于在非均相条件下,催化剂和CSA很难进行链交换反应,因此聚合是在均相条件下发生的,一般均相聚合的温度大于120 ℃,这就要求催化剂和链穿梭剂具有很好的耐热性;(2)催化剂和CSA之间要有良好的匹配,CSA与催化剂之间链交换反应速率应大于链终止反应速率,即聚合物链在终止前能与CSA至少完成一次交换;(3)不同活性中心具有不同的选择性(立体选择性,单体插入能力不同),例如上述过程中共聚单体对Cat.1和Cat.2上活性中心的插入能力不同。
2 链穿梭聚合的研究进展
自“链穿梭聚合”概念提出以来,迅速引起国内外科研工作者的极大关注。鉴于陶氏化学公司“链穿梭聚合”体系较严苛的催化剂匹配原则以及链穿梭剂筛选条件,人们开始致力于开发新的催化剂,采用不同的聚合方法来研究链穿梭聚合并取得明显进展。
2.1 基于链行走的链穿梭聚合
1995年Brookhart等[11]6415提出α-二亚胺镍/钯催化剂通过链行走的方式,可以在常温常压下得到支化度较高的无定形态聚乙烯。于是,人们根据这一重大发现将其应用于链穿梭聚合中并取得了一些研究成果。
Wang等[24]仅采用乙烯单体,在链穿梭剂的存在下,通过链行走和链穿梭聚合,制备了一种新型的线性-超支化多嵌段共聚物。选取Cat.3、Cat.4(结构式见图3)与CSA组成的催化剂体系,茂金属催化剂(以下简称MAO)为助催化剂,在20 ℃下催化乙烯聚合。在Cat.3与MAO的作用下,乙烯会发生链行走,产生支链,Cat.4则产生线形聚乙烯,在链穿梭剂的作用下,两条不同的链段在两个活性中心之间交替增长,从而成功制备线形-超支化嵌段共聚物。所制得的嵌段共聚物熔点在120 ℃以上,链穿梭剂用量较大时,熔融焓低至2.5 J/g,并且嵌段共聚物的相对分子质量分布为1.91~2.21,符合Schulz-Flory分布。
Martins等[25]用一种新型的耐热性较好的α-二亚胺镍催化剂(Cat.5结构式见图3)和二茂锆催化剂(Cat.6结构式见图3)组成的催化体系,在链转移剂CSA的存在下,分别在60 ℃、80 ℃、100 ℃的条件下,催化乙烯均聚形成“软”、“硬”相间的嵌段共聚物。其中,Cat.5在较高温度下催化乙烯形成高度支化聚乙烯,Cat.6催化乙烯形成具有较高熔融温度的线形聚乙烯。嵌段共聚物的熔点在125 ℃左右,并且随着链穿梭剂用量的增加或者聚合温度升高,聚合物的熔点和结晶度都略有增加。
Xiao等[26]选用了两种不同类型的α-二亚胺镍催化剂(Cat.3~Cat.7结构式见图3),以CSA作为链转移剂,MAO作为助催化剂,在20 ℃下于常压下催化乙烯均聚。利用α-二亚胺镍在催化乙烯时具有链行走的特性,Cat.3催化乙烯可形成支化度大于100的高支化度的聚乙烯链段,Cat.7在同样的条件下可催化乙烯形成中等支化度的聚乙烯链段,在链穿梭剂的作用下,两条支化度不同的链段在两个催化剂上交替增长,形成一种新型的支化-高度支化的嵌段聚烯烃。当n(Zn)/n(Cat.3+Cat.4)=150时,嵌段共聚物包含50.8%的高度支化聚乙烯和49.2%的中等支化度聚乙烯,相对分子质量分布由双峰分布变为单峰分布,且Mw/Mn为2.50。

图3 链穿梭聚合选用的Cat.3~Cat.7
2.2 过渡金属催化剂参与的链穿梭聚合
Busico等[27]7736创造性地将链穿梭的概念扩展到合成等规立构嵌段聚丙烯的研究领域。催化体系由(吡啶基-酰胺)二甲基铪配合物(Cat.8结构式见图4)、MAO以及MAO游离的三甲基铝组成。选择Cat.8作为主催化剂,一方面是由于对映位点的控制,Cat.8对丙烯具有高度全同立构选择性;另一方面该催化剂是外消旋的,这就可以用对映异构体的形式来使用它,换言之,对外消旋催化剂而言,插入单体的手性与聚丙烯链相邻结构单元的手性关系仅由催化剂活性中心决定。聚合过程中,在链穿梭剂三甲基铝的作用下,在两个手性相反的活性中心生长的聚合物链段发生无规的交换,就会产生对应面不同的等规立构嵌段聚丙烯。当使用单一的对映体为催化剂时,观察不到立体嵌段结构的存在,这也证明了链穿梭聚合的发生。
Tynys等[28]选取两种不同的二茂锆催化剂(Cat.9、Cat.10结构式见图4),以MAO为助催化剂,在不同浓度的三甲基铝条件下,催化丙烯均聚形成立构嵌段聚丙烯。其中Cat.9可催化丙烯形成相对分子质量较高的间规聚丙烯,Cat.10则催化丙烯形成相对分子质量较低的等规聚丙烯。聚合过程中,分子链在不同的活性中心和三甲基铝间大量转移,形成嵌段共聚物。并且随着三甲基铝浓度的变化,催化剂的活性、聚合物的相对分子质量、熔融行为、微观结构都会发生变化。
Wang等[29]选用两种具有不同立构选择性的双元茂金属催化体系(Cat.4结构式见图3,Cat.11结构式见图4),以MAO为助催化剂,CSA为链穿梭剂催化丙烯均聚。其中,Cat.4可作为等规立构催化剂前体,Cat.11作为无规催化剂前体催化丙烯聚合,利用链穿梭剂使增长链在两个活性中心交叉增长。加入适量的CSA后,共聚物熔点达到125 ℃,结晶度为22%。与不添加CSA的样品对比,共聚物的核磁谱图中,出现无规结构聚丙烯的特征峰,也证明了链穿梭反应的发生。
Tritto等[30]将链穿梭聚合应用到乙烯与环烯(降冰片烯)的共聚。选用两种茂金属催化剂Cat.11和Cat.12(结构式见图4),这两种催化剂对单体插入能力选择性不同,Cat.11具有较好的共聚能力,生成的聚合物玻璃化转变温度高达195 ℃,Cat.12共聚能力较弱,生成聚合物玻璃化转变温度约为130 ℃。在两种催化剂和链穿梭剂共同存在的体系中,聚合物的玻璃化转变温度呈单峰分布,并且根据CSA加入量的不同,嵌段共聚物玻璃化转变温度可在140~190 ℃之间调节。

图4 链穿梭聚合选用的Cat.8~Cat.13
2.3 稀土金属催化剂参与的链穿梭聚合
前面所提到的催化体系除Busico[27]7737选取三甲基铝作为催化剂之外,其它体系都是用CSA作为链穿梭剂。除此之外,三异丁基铝[31]、烷基镁[32]等烷基金属也可以作为链穿梭剂应用到链穿梭聚合中。
PAN等[31]再次将链穿梭聚合引入一个全新的领域——稀土金属催化剂催化共轭双烯均聚或与其它单体共聚。该催化体系包括三种Sc系催化剂:Cat.13、Cat.14、Cat.15(结构式见图5),选取三异丁基铝作为链穿梭剂,在常温常压下催化异戊二烯、苯乙烯、丁二烯共聚。Cat.13对苯乙烯显示出较高的催化活性和立体选择性,而对异戊二烯催化活性较低;Cat.14对异戊二烯和丁二烯具有较高的催化活性,并且可催化形成顺式-1,4结构的聚异戊二烯和聚丁二烯;Cat.15对异戊二烯显示出较高的3,4-结构选择性。选取Cat.13与其它两种Sc系催化剂组合,在链穿梭剂的作用下可得到不同立体构型的嵌段共聚物。例如:选取Cat.13和Cat.14,加入三异丁基铝,在25 ℃条件下制备的间规聚苯乙烯与顺-1,4-聚异戊二烯的嵌段共聚物,熔点接近270 ℃,与间规聚苯乙烯熔点(272 ℃)接近;玻璃化转变温度接近-60 ℃,与聚异戊二烯玻璃化转变温度(-62 ℃)接近。在相同的聚合条件下,加入丁二烯,可得到间规聚苯乙烯、顺-1,4-聚异戊二烯和顺-1,4-聚丁二烯的三嵌段共聚物,熔点与玻璃化转变温度都与上述的两嵌段共聚物相近。当选取Cat.13和Cat.15作为催化剂时,可得到间规聚苯乙烯和3,4-聚异戊二烯嵌段共聚物,熔点约为260 ℃,与间规聚苯乙烯熔点接近;玻璃化转变温度约为30 ℃,与1,2-聚异戊二烯玻璃化转变温度(40 ℃)接近。
Valente等[32]在2014年报道,利用链穿梭聚合直接合成“软”、“硬”相间无规嵌段共聚物。选取Cat.16和Cat.17(结构式见图5)作为主催化剂,正丁基乙基镁作为链穿梭剂,催化异戊二烯和苯乙烯共聚,形成反-1,4-异戊二烯和苯乙烯的嵌段共聚物。共聚物的玻璃化转变温度可以通过改变单体的配比和链穿梭剂的浓度进行调控。从SAXS和AFM图上可知,得到的产物可通过自组装的方式形成纳米结构,从而得到较硬的链段。
Liu等[33]通过仅使用一种单体异戊二烯,以稀土金属催化剂(Cat.18、Cat.19结构式见图5)为主催化剂,通过链穿梭聚合制备了一种新型材料。这两种稀土金属催化剂对异戊二烯具有不同的立体结构选择性,其中Cat.18可催化异戊二烯形成反-1,4-聚异戊二烯,Cat.19则对聚异戊二烯显示出较高的3,4-结构选择性,并且两种稀土金属催化剂在三异丁基铝的作用下都显示出较高的链转移效率。基于这一现象,在体系中同时加入两种催化剂,在链穿梭剂的作用下,得到了反-1,4-异戊二烯和3,4-异戊二烯的嵌段共聚物。共聚物的相对分子质量分布在2.0左右,玻璃化转变温度在-38~2 ℃内可调。
Zinck等[34]在2017年也提出仅使用异戊二烯这一种单体,利用链穿梭聚合制备反式、顺式交替排列的立构多嵌段聚异戊二烯。催化体系主要包括两种具有不用立体选择性的稀土金属催化剂(Cat.16和Cat.20结构式见图5),烷基铝做链穿梭剂,在50 ℃下催化异戊二烯聚合。Cat.16催化单体聚合可得到反-1,4-聚异戊二烯,在室温条件下是半结晶的,可充当嵌断共聚物中的“硬段”;而Cat.20催化单体得到顺-1,4-聚异戊二烯,室温下为无定形状态,成为嵌段共聚物中的“软段”。所得立构嵌段共聚物的相对分子质量分布为2.58,玻璃化转变温度为-66.8 ℃,熔点可达到38.6 ℃。
Li等[35]利用一种三元催化体系Nd(CF3SO3)3·H2O·3TBP/Mg(n-Bu)2/MMAO(其中TBP为磷酸三丁酯;MMAO为改性甲基铝氧烷)催化丁二烯聚合,得到顺式-1,4和反式-1,4-聚丁二烯的多嵌段共聚物。共聚物中顺式-1,4和反式-1,4聚丁二烯的含量、相对分子质量及其分布都是可以通过反应条件控制的。在该反应中,Mg(n-Bu)2既可作为助催化剂,又能起到链穿梭剂的作用,与主催化剂共同作用可催化单体生成相对分子质量分布较窄的反式-1,4-聚丁二烯;而主催化剂在MMAO的作用下,可催化单体形成相对分子质量较高的顺式-1,4-聚丁二烯。与之前提及的链穿梭聚合体系中,由主催化剂控制单体的立体选择性或单体插入顺序不同,在这项工作中,作者首次尝试通过助催化剂来调节单体的立体选择性,这无疑打破了人们对传统“链穿梭聚合”概念的认识,更加扩大了链穿梭聚合的应用领域。
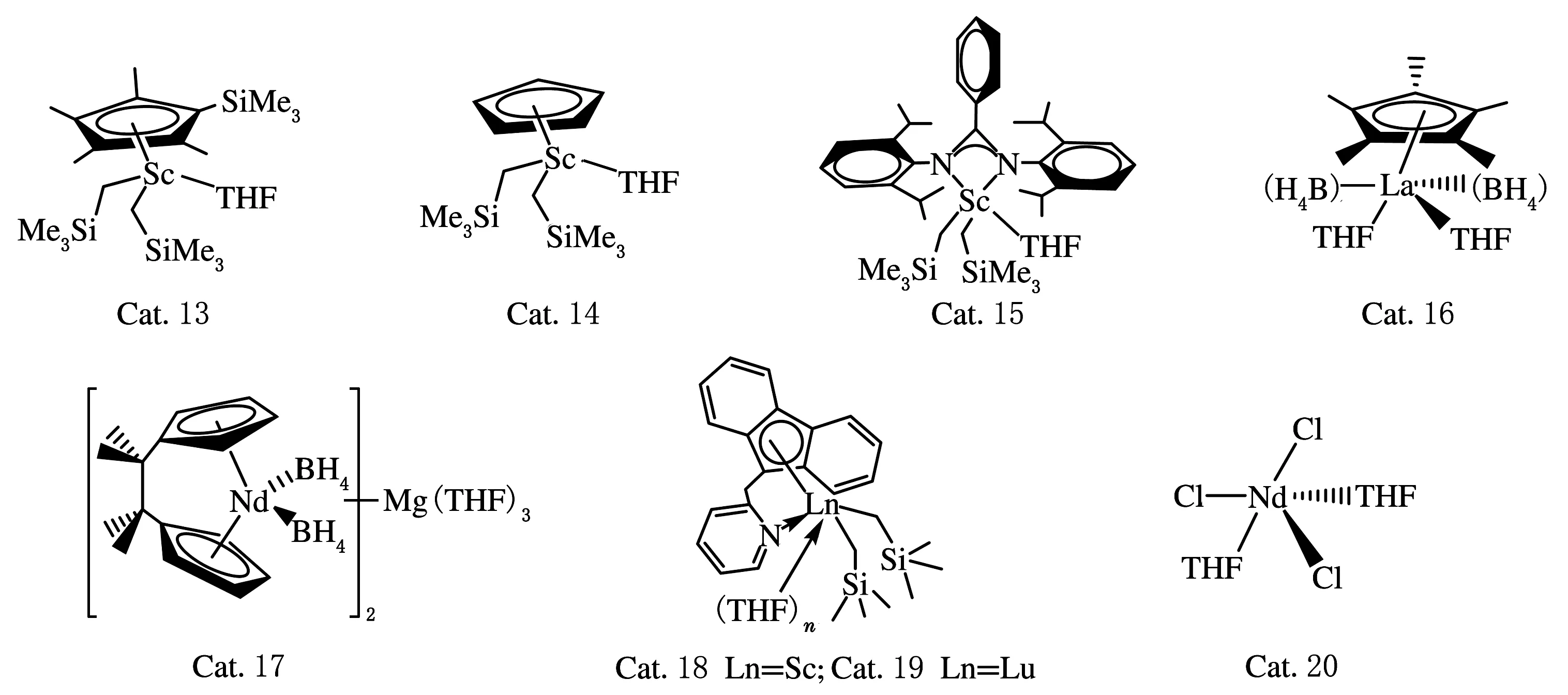
图5 链穿梭聚合选用的Cat.13~Cat.20
3 结束语
每一次新材料的出现都会极大地推动人类社会的进步,提高人们的生活水平。新型聚合物和合成聚合物的新方法是高分子化学前沿的永恒主题。通过链穿梭聚合得到的共聚物的相对分子质量分布一般在2.0左右,并且可以通过改变单体配比、催化剂种类、反应条件等调节共聚物的微观结构,从而获得更加优异的性能,具有广阔的应用前景。通过链穿梭聚合的方法制备热塑性弹性体不仅能降低生产成本,简化生产工艺及流程,还能扩大单体的使用范围,制备更多的符合要求的产品,这些都表明,链穿梭聚合具有非常光明的工业化前景。但鉴于链穿梭聚合体系中两种催化剂和单体匹配难度较大,确保聚合物链在终止前能与CSA至少完成一次交换的苛刻条件,链穿梭聚合的发展还是比较缓慢的。如何设计更加新颖的催化剂来降低筛选的难度,缩短生产周期,如何找出链转移效率更高、与催化剂匹配性更好的链穿梭剂,如何找到一个能够判断链穿梭反应是否发生的简单测试方法来提高生产效率,这些都是值得科学家进一步尝试的研究工作。
参 考 文 献:
[1] ZIEGLER K,HOLZKAMP E,BREIL H,et al.Themulheim normal pressure polyethylene process[J].Angew Chem,1955,67(19/20):541-547.
[2] NATTA G.Stereospecific catalysts and isotactic polymers[J].Angew Chem,1956,68(12):393-403.
[3] KOO C M,WU L F,LIM L S,et al.Microstructure and mechanical properties of semicrystalline-rubbery-semicrystalline triblock copolymers[J].Macromolecules,2005,38(14):6090-6098.
[4] DIETRICH U,HACKMANN M,RIEGGER B,et al.Control of stereoerror formation with high-activity “dual-side” zirconocene catalysts:A novel strategy to design the properties of thermoplastic elastic polypropenes[J].J Am Chem Soc,1999,121(18):4348-4355.
[5] 张志德,陈玉琴,余立新,等.茂金属催化剂及其应用[J].山东化工,1996(3):36.
[6] CHUM P S,KAO C I,KNIGHT G W.Structure-property relationships inpolyolefins made by constrained geometry catalyst technology[J].Plast Eng,1995,51(6):21-23.
[7] MICHAEL A G,MORIS S E,TOBIN J M,et al.Chiral,non-C2,symmetric zirconocene complexes as catalysts for stereoregular a-olefin polymerization[J].J Am Chem Soc,1993,115(8):3326-3327.
[8] PAUL A D,TOBIN J M.Cationic metallocene olefin polymerization catalysts-thermodynamic and kinetic-parameters for ion-pair formation,dissociation,and reorganization[J].J Am Chem Soc,1995,117(22):6128-6129.
[9] PAUL A D,COLIN L B,TOBIN J M.Highly electrophilic olefin polymerization catalysts.Quantitative reaction coordinates for fluoroarylborane/alumoxane methide abstraction and ion-pair reorganization in group 4 metallocene and “constrained geometry” catalysts[J].J Am Chem Soc,1998,120(8):1772-1784.
[10] COATES G W,WAYMOUTH R M.Oscillating stereo-control:a strategy for the synthesis of thermoplastic elastomeric polypropylene[J].Science,1995,267(5195):217-219.
[11] JOHNSON L K,KILLIAN C M,BROOKHART M.New Pd(Ⅱ)-and Ni(Ⅱ)-based catalysts for polymerization of ethylene and alpha-olefins[J].J Am Chem Soc,1995,117(23):6414-6415.
[12] KILLIAN C M,TEMPEL D J,JOHNSON L K,et al.Living polymerization ofα-olefins using NiⅡ-α-diimine catalysts.Synthesis of new block polymers based onα-olefins[J].J Am Chem Soc,1996,118(46):11664-11665.
[13] GATES D P,SVEJDA S A,ONATE E,et al.Synthesis of branched polyethylene using(α-diimine) nickel(Ⅱ) catalysts:influence of temperature,ethylene pressure,and ligand structure on polymerproperties[J].Macromolecules,2000,33(7):2320-2334.
[14] CAMACHO D H,SALO E V,ZILLER J W,et al.Cyclophane-based highly active late-transition-metal catalysts for ethylene polymerization[J].Angew Chem Int Ed,2004,43(14):1821-1825.
[15] RHINEHART J L,BROWN L A,LONG B K.A robust Ni(Ⅱ)α-diimine catalyst for high temperature ethylene polymerization[J].J Am Chem Soc,2013,135(44):16316-16319.
[16] ARRIOLA D J,CARNAHAN E M,HUSTAD P D,et al.Catalytic production of olefin block copolymers via chain shuttling polymerization[J].Science,2006,312(5774):714-719.
[17] DOMSKI G J,ROSE J M,BROOKHART M,et al.Livingalkene polymerization:New methods for the precision synthesis of polyolefins[J].Prog Polym Sci,2007,32(1):30-92.
[18] GIBSON V C.Shuttling polyolefins to a new materials dimension[J].Science,2006,312(5774):703-704.
[19] ZINCK P,VALENTE A,MORTREUX A,et al.In situ generated half-lanthanidocene based catalysts for the controlled oligomerisation of styrene:Selectivity,block copolymerisation and chain transfer[J].Polymer,2007,48(16):4609-4614.
[20] KEMPE R.How to polymerize ethylene in a highly controlled fashion[J].Chem Eur J,2007,13(10):2764-2773.
[21] AMIN S B,MARKS T J.Versatile pathways for in situ polyolefin functionalization with heteroatoms:Catalytic chain transfer[J].Angew Chem,2008,47(11):2006-2025.
[22] 王凤,张贺新,张学全,等.双烯烃配位链转移聚合研究进展[J].高分子通报,2013(5):57-64.
[23] ZINTL M,RIEGER B.Novel olefin block copolymers through chain-shuttling polymerization[J].Angew ChemInt Ed,2007,46(3):333-335.
[24] XIAO A G,WANG L,LIU Q Q,et al.A novel linear-hyperbranched multiblock polyethylene produced from ethylene monomer alone via chain walking and chain shuttling polymerization[J].Macromolecules,2009,42(6):1834-1837.
[25] ROBERTO M,LETICIA Q,GIULIANA S,et al.Polymerization of ethylene with catalyst mixture in the presence of chain shuttling agent[J].Chemistry&Chemical Technology,2012,6(2):153-162.
[26] XIAO A G,ZHOU S B,LIU Q Q.A novel branched-hyperbranched block polyolefin produced via chain shuttling polymerization from ethylene alone[J].Polymer-Plastics Technology and Engineering,2014,53(17):1832-1837.
[27] ALFANO F,BOONE H W,BUSICO V,et al.Polypropylene “chain shuttling” atenantiomorphous and enantiopure catalytic species:direct and quantitative evidence from polymer microstructure[J].Macromolecules,2007,40(22):7736-7738.
[28] TYNYS A,EILERTSEN JL,SEPPALA JV,et al.Propylene polymerizations with a binarymetallocene system-chain shuttling caused by trimethylaluminium between active catalyst centers[J].J Polym Sci,Polym Chem,2007,45(7):1364-1376.
[29] XIAO A G,WANG L.Propylene polymerization catalyzed by rac-Et(Ind)2ZrCl2/Cp2ZrCl2in the presence of ZnEt2[J].Designed Monomers and Polymers,2009,12(5):425-431.
[30] INCORONATA T,LAURA B,GIULIA S,et al.Novelnorbornene copolymers with transition metal catalysts[J].J Organomet Chem,2015,798(2):367-374.
[31] PAN L,ZHANG K Y,NISHIURA M,et al.Chain-shuttling polymerization at two different scandium sites:regio-and stereospecific “one-pot” block copolymerization of styrene,isoprene and butadiene[J].Angew Chem,2011,123(50):12218-12221.
[32] VALENTE A,STOCLET G,BONNET F,et al.Isoprene-styrene chain shuttling copolymerization mediated by a lanthanide half-sandwich complex and alanthanidocene:straightforward access to a new type of thermoplastic elastomers[J].Angew Chem Int Ed,2014,53(18):4638-4641.
[33] LIU B,CUI D M.Regioselective chain shuttling polymerization of isoprene:an approach to access new materials from single monomer[J].Macromolecules 2016,49(17):6226-6231.
[34] PHUPHUAK Y,BONNET F,ZINCK P,et al.Isoprene chain shuttling polymerizationbetweencis and trans regulating catalysts:straightforward access to a new material[J].Chem Commun,2017,53(38):5330-5333.
[35] DAI Q Q,ZHANG X Q,LI Y Q,et al.Regulation of the cis-1,4-and trans-1,4-polybutadiene multiblock copolymers via chain shuttling polymerization using a ternary neodymium organic sulfonate catalyst[J].Macromolecules,2017,50(20):7887-7894.
猜你喜欢
煤炭与化工(2021年6期)2021-08-06 10:04:10
Atmospheric and Oceanic Science Letters(2016年4期)2016-11-23 03:30:18
安徽大学学报(自然科学版)(2016年2期)2016-09-20 12:09:27
吉林农业(2016年15期)2016-08-30 03:45:48
吉林农业(2016年8期)2016-05-14 13:52:43
橡塑技术与装备(2016年21期)2016-02-25 06:33:12
石油化工(2015年9期)2015-08-15 00:43:05
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:53
有机氟工业(2014年4期)2014-06-01 12:30:37
弹性体(2014年1期)2014-05-21 02:05:37
