
普通油茶花发育过程数字表达谱测序及分析
2018-06-05 01:08胡玉玲姚小华任华东王开良张山
西部林业科学 2018年3期
胡玉玲,姚小华,任华东,王开良,张山
(1.铜仁学院农林工程与规划学院,贵州 铜仁 554300;2.湖南应用技术学院, 湖南 常德 415000;3.中国林业科学研究院亚热带林业研究所, 浙江 杭州 311400)
高等植物开花是植物生命活动的重要现象,这不仅关系着该物种的延续,而且与农业生产密切相关[1]。植物成花的整个过程中较关键的是成花诱导、花芽分化和花器官形成,这些过程直接决定了植物生育周期长短,与植物的产量及品质关系紧密[2]。油茶(CamelliaoleiferaAbel.)是典型多花而不高产的物种,一方面由于油茶大部分品种自交可育性低,另一方面,油茶生产周期接近1年,茶果经历了4个物候差异非常明显的季节,特别是秋冬季低温潮湿和夏季的高温少雨。因此培育出自交可育性高的油茶品种是当务之急,由于油茶是多年生木本植物,育种周期较长,可见培养优良新品种非一蹴而就的事情。有研究表明,油茶花期遇到寒冷潮湿天气会严重影响油茶产量,同时冬天温度过低也会影响油茶幼果过冬从而影响油茶产量[3]。
基因工程是较有效的植物遗传改良方式,尤其对多年生木本植物。针对上述影响油茶产量限定因子实施基因工程,需要明确并解决如下几个问题:首先控制油茶成花转变的关键基因,以有效实现人工成花转变,缩短育种周期;其次控制花器官形成基因,如与雄蕊发育有关基因,以人工获得雄性不育系;再次影响花开放进程相关基因,以改变油茶花期或者通过栽培措施改变花期以避开不利天气,其中揭示油茶花发育分子机制最为重要。所有生物的整个生命活动就是不同编码蛋白的基因进行选择性开启和关闭结果,为准确获得需要遗传改良成花相关基因,必须了解基因表达过程。细胞的特定发育阶段,一般只有15%的基因进行了表达,这种在生物个体发育的不同阶段、不同组织器官中,按照一定时空顺序,基因的有序表达过程,就是基因的差别表达(Differential expressing)[4-5]。采用Solexa技术对植物样本中数以百万计的cDNA标签进行序列测定,能够对整个转录组和表达谱进行数字化的分析,数字的列信号可以准确、特异地反映对应基因的真实表达情况。这种技术甚至可以精确地检测低至一两个拷贝的稀有转录本,并精确定量高达十万个拷贝的转录本的表达量变化。由于序列无需事先设计,数字表达谱数据具有极佳的实时性,可以充分利用当前爆发式增长的信息资源,并与未来研究相衔接,数字表达谱可以检测到许多未曾注释的基因和基因组部位,为新基因的发现提供了良好的线索,然后通过生物信息学的分析及样品间基因表达的比对可以初步判定基因表达过程[6-8]。
因此,本研究选择普通油茶花芽分化前、萼片形成期、花瓣形成期、雌雄蕊形成期、子房及花药形成期和雌雄蕊成熟期的花芽作为试验材料,对该6个时期样品进行表达谱测序并进行分析,以期揭示普通油茶花发育过程的分子机制及成花关键基因,为培育自交可育性高的油茶品种提供强有力的理论依据。
1 试验材料与方法
1.1 试验材料
普通油茶长林4号(Camelliaoleifera‘ChangLin 4’)优良无性系。
1.2 采样方法
根据相关文献报道[9-11],分别在油茶花芽分化前(5月8日),萼片形成期(5月28日)、花瓣形成期(6月15日)、雌雄蕊形成期(7月3日)、子房和花药形成期(7月25日)和雌雄蕊成熟期(8月25日)采集花芽,样品采集后都在液氮带回保存,放入-80℃的超低温冰箱中保存备用。
1.3 RNA提取
RNA提取采用艾德莱公司EASYspin Plus植物RNA快速提取试剂盒(目录号为RN38)使用手册进行,采用微量分光光度计Nanodrop 2000及琼脂糖凝胶电泳检测RNA质量,保证RNA样品浓度400ng/μL,28S︰18S大于1.8。
1.4 上机测序前准备和方法
利用富含ploy(T)低吸附磁珠对Total RNA中的mRNA进行富集纯化处理,在高温条件下,利用2价阳离子打断mRNA以选取合适大小的目的片段,利用反转录酶和随机引物将打断后mRNA片段反转录形成cDNA第一条链,然后加入缓冲液、dNTPs,在RNase H和DNA polymerase I的作用下合成第二条cDNA链。对反转录合成的双链cDNA进行末端修复、3’末端加A、连接接头,通过琼脂糖凝胶电泳,筛选一定范围大小的片段。PCR扩增,上机测序。
1.5 生物信息学分析
用The BLAST-Like Alignment Tool对参考基因序列比对,基因表达定量分析,表达量用RPKM(Reads Per kb per Million reads)表示,RPKM值的计算公式,RPKM=(map the gene reads)/〔map all gene reads(million)×the gene length(kb)〕。
用IDEG6进行差异表达分析,用Cluster 3.0[12-13]进行基因聚类分析,从参考基因注释信息里面提取出差异表达基因的信息,同时对差异表达基因做GO和KEGG Pathway的显著性富集分析。通过GO功能显著性富集分析能确定差异表达基因行使的主要生物学功能;通过Pathway显著性富集能确定差异表达基因参与的主要生化代谢途径和信号转导途径。整个过程见图1。

图1 数字表达谱生物信息分析流程图
2 结果与分析
2.1 表达谱测序
2.1.1 测序数据统计
采用Solexa HiSeq 2000对每个样品进行单端测序,统计测序数据产出、数据质量以及cycleQ20%等情况。从表1可以看出6个发育时期的测序数据数量都超过8M,完全满足后续的分析,E2样品的GC含量最低(45.19%),E5样品的GC含量最高(54%),测序的cycleQ20%都达到100%,保证测序结果准确性。

表1 测序数据
注:E1为 5月9日,E2为7月3日,E3为8月25日,E4为5月29日,E5为6月15日,E6为7月25日,下同。
2.1.2 各样品质量评估及相关基因表达特点
测序数据质量评估分别有cDNA片段随机性基因和测序饱和度分析。评价mRNA打断随机程度一般是以reads在参考基因上的分布情况来确定的。由于不同参考基因有不同的长度,必须先标准化reads在参考基因上的位置(reads在基因上的位置与基因长度的比值),然后统计基因的不同位置比对上的reads数。如果打断随机性好,reads在基因各部位应分布得比较均匀。从图2 cDNA打断可以看出,测序的reads数没有3’或5’严重的偏向性,就可以保证结果的准确。

E1E4E5E2E6E3
图2cDNA片段随机性检验
Fig.2 Random testing of cDNA fragments
样品中测序饱和度分析是用来衡量一个样品的测序量多少的标准,随着测序量(reads数量)的增多,检测到的基因数也随之上升,当测序量达到某个值时,其检测到的基因数增长速度趋于平缓,说明检测到的基因数趋于饱和。从图3可以看出,各个样品中检测到的基因数基本趋于饱和,说明6个样品的测序量已经覆盖了绝大部分表达的基因,基本可以挖掘全部的差异基因。

E1E4E5E2E6E3
图3测序饱和度分析
Fig.3 Saturation analysis of sequencing
基因表达定量分析是根据各样品比对到参考基因序列上的reads数,来计算各样品的基因表达丰度信息,利用RPKM值来反映样品的基因表达丰度(图4)。基因表达丰度分布分析是根据各样品中基因的RPKM值对各基因的表达丰度进行估算,从整体水平了解样品中基因的表达模式。一般来说,不均一性、冗余性是细胞中mRNA的显著特征,少量种类的基因表达丰度极高,而大部分种类的基因表达水平很低甚至极低。从6个样品的表达基因中,RPKM值>100的高表达基因数约占总基因数量的1.5%,而此区间内基因的总表达丰度约占全部基因表达丰度的75%;从基因表达量分布图(图5)可以看出,6个样品中基因表达的RPKM值在0-5区间内的低表达基因数约占总基因数量的 70%,而此区间内基因的总表达丰度约占全部基因表达丰度的5%;其表达模式符合 mRNA不均一性、冗余性的特征。从图5中的E5和E6可以看出RPKM值小于100的非常少,绝大部分基因表达量都大于100。

E1E4E5E2E6E3
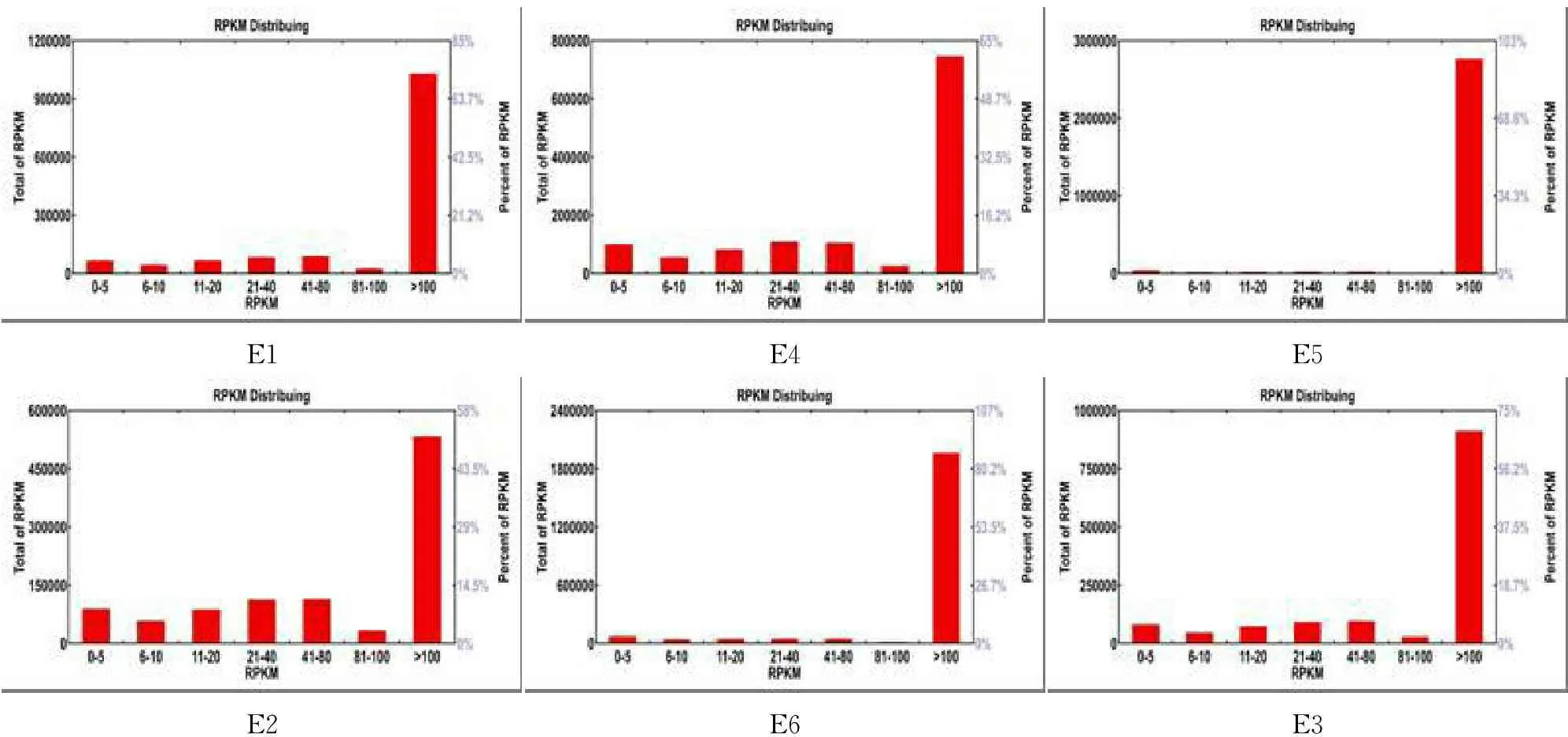
图4 基因表达分布图
图5基因表达量分布图
Fig.5 Distribution of gene expression level
2.1.3 比对效率统计
由于参考基因序列是转录组部分构建的Unigenes库序列,表达谱测序的样品通过统计比对效率,可以检查数据是否正常,这也是后续基因表达及差异分析的结果准确性的保证。通过使用Blat比对软件,将各样品测序得到的reads分别与参考基因序列进行比对,样品的Mapped percentage(表2)均超过60%,因此可以判定比对效率较高,测序结果可以进行后续分析。
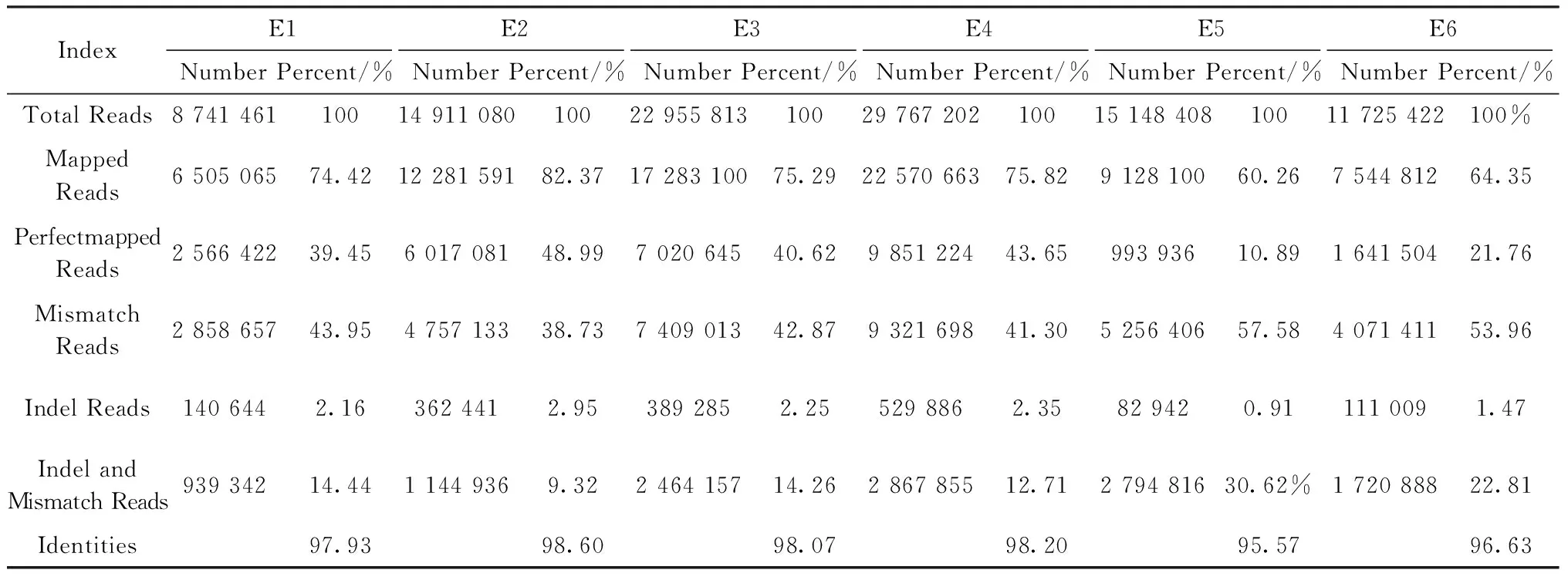
表2 样品与参考基因的比对效率
2.2 差异表达基因分析
差异表达基因分析见表3。

表3 两两样品间比较差异表达基因数量统计
从表3可以看出,5月9日(E1)和8月25日(E3)的差异表达基因明显较少,可见这两个时期油茶都在进行最基本并相近的生命活动,各个时期和7月3日(E2)与6月15日(E5)差异基因最多,其中上调基因也最多,达到19 408个,其次是5月29日(E4)与6月15日(E5),E4对E5的上调基因有16 644,下调仅107个,可见这两个时期生命代谢活动最为活跃。
2.3 差异表达基因生物信息学分析
根据测序Reads与参考基因序列的比对,得到对应基因在样品中的表达丰度,基因在不同样品间的表达丰度的不同,寻找差异表达的基因。结合差异基因的功能注释,进行pathway(KEGG)与GO的富集分析,以及对差异表达基因进行模式聚类的分析。富集分析采用fisher精确检验,通过Bonferroni校正法进行校正,得到差异基因显著富集的pathway和GO功能类,以便进行进一步的分析研究。
2.3.1 花发育6个时期样品差异基因聚类分析
根据基因表达丰度,使用cluster聚类软件对所有样品间的差异表达基因做层次聚类分析,将具有相同或相似表达行为的基因进行聚类。从图6各列聚类结果可以看出,在所有的差异基因中绝大部分基因表达行为相似,只有少部分基因扮演着关键的生物功能。从不同样品聚类来看,E1、E3(5月9日和8月25日)和E5、E6(6月15日和7月25 日)基因表达水平较接近,E2、E5(6月15日和7月3日)基因表达水平差异最为明显。

图6 差异基因聚类图
注:图中列代表不同的样品;行代表不同的基因;相应的颜色代表基因在样品中表达丰度的高低。黄色表示基因在样品中高表达,蓝色表示基因在样品中低表达
Fig.6 Differences in gene clustering
2.3.2 花发育6个时期样品差异表达基因生物功能分析
从图7-A可以看出,执行细胞组成功能中细胞外基质和细胞核与差异基因关系紧密;分子功能中核酸和蛋白绑定转录因子活性与差异基因关系紧密;生物学过程中的抗氧化性,细胞增殖,病毒繁殖和各种生命运动与差异基因关系紧密。
从图7-B可以看出,现较多基因的生命功能有翻译,核糖体结构和生物合成为245条,转录因子为372条,复制,重组和修复为320条,细胞信号转导机制为285条,翻译后修饰,蛋白质周转,伴侣蛋白为283条,能源生产和转换为114条,碳水化合物运输和代谢为136条,氨基酸的转运和代谢为122条。
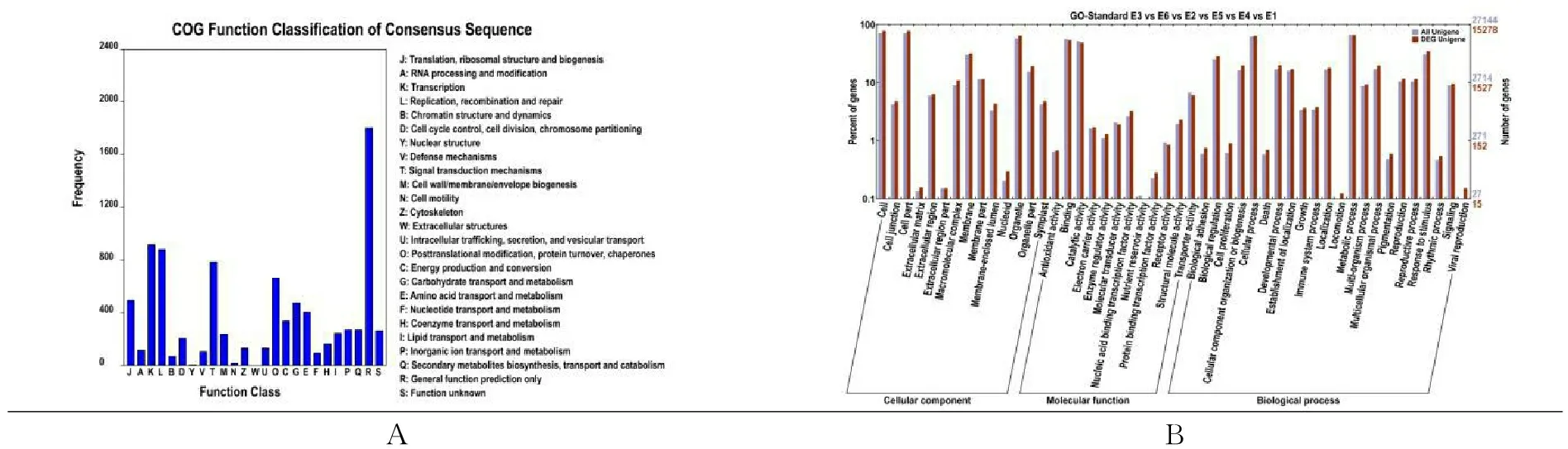
AB
图7差异基因生物功能分析
注:A为GO的注释图,B为Cog分类图
Fig.7 Biological attributes analysis on differential gene
2.4 花器官形成相关unigenes表达
花器官形成是植物成花的重要过程,ABC模式是目前花器官形成最成熟的理论,从表4可以看出,A类基因Unigene数量最多,其中Unigene67783在不同的发育阶段差异较明显,表达水平也较高;B类基因有9条Unigene,其中Unigene56059 表达水平较高,各发育阶段差异也较明显;属于C类基因的Unigene数量有9条,总体表达都不高,差异不明显。
3 结论与讨论
3.1 数字表达谱测序
数字基因表达谱测序方法是直接测定每个基因特异性的表达标签序列,以计数表达标签序列数目来确定该基因的表达量,从而大大提高了定量分析的准确性[14-15]。采用Solexa方法测序,对于那样表达差异不大的基因,也能够检测其表达的差异性,同时也能够发现新的转录本或者基因组表达调控区域等。另外该技术不用事先设计探针,采用直接测序的方式,因此即使不了解物种基因信息,也可以直接对该物种进行测序,包括未知基因在内的全基因组表达谱分析。从本文来看,从花发育的6个时期样品测序reads与转录组部分测序构建的Unigene库的序列对比[16],对比效率都超过60%,通过生物信息学的分析更有效地了解花发育的基因表达。本次试验采样严格把握花发育的不同阶段,在高通量转录组测序数据基础上,进行表达谱测序,本文表达谱测序数据量最少者超过8M,最多数据量近30M。因此,表达谱数据基本覆盖了所有表达标签,对准确后续研究及生物信息学分析奠定了基础,为有效及准确分析基因表达提供了保障,通过样品间两两比较获得了15组的比较数据,同时得到差异表达基因26861个,对差异基因生物信息学的分析为有效获得成花相关基因建立重要基础。

表4 花器官形成相关Unigenes
3.2 生物信息学分析
普通油茶的成花决定机理是进行花期调控和缩短花期研究的重要依据,对加速育种周期及解决花期不遇有重要意义。对花器官形成的研究同样也非常重要,一方面通过了解相关因素及基因对花器官影响机制,可以为研究雄性不育及人工调控、花色及形态及亲和性研究提供重要依据。AP1和AP2属于MADS-box基因家族,已经证明不仅和植物花器官形成有关,同时也与植物花芽分化关系密切[17-18]。从本文分析可以看出,AP1在花芽分化前期表达量较高,AP2在花芽分化期及花器官形成后期表达较突出,结合之前研究的FT基因表达结果,FT基因促进AP1和AP2基因表达,而不经LFY基因。通过本文分析PI基因与雄蕊发育关系有关十分密切,这与花器官发育的ABC模型一致[19-21],通过研究发现雄蕊形成期7月3日与花瓣形成期6月15日差异基因最多,上调基因达到19 408个,可见6月份到7月份是花器官形成的关键时期,通过对该时期基因调控可以有效控制花器官发育,如获得单性花植株,观赏性好的油茶。
参考文献:
[1] 陈晓亚,汤章城.植物生理与分子生物学[M].北京:高等教育出版社,2007.
[2] Paul K B,Ruth M B,Joshua S M,etal.Multiple pathways in the decision to flower:enabling,promoting,and resetting[J].The Plant Cell Online,2004,16(suppl 1):18-31.
[3] 曾燕如,黎章矩,戴文圣.油茶开花习性的观察研究[J].浙江农林大学学报,2009,26(6):802-809.
[4] Martin C,Yuan T,Ghia E,etal.Green fluorescent protein as a marker for gene expression[J].Science,1994,263(5148):802-805.
[5] 宁顺斌,王玲.几种差别表达基因显示技术及其在植物方面的应用[J].生命科学,1999,11(3):140-143.
[6] Morrissy A S,Ryan D M,Allen D,etal.Next-generation tag sequencing for cancer gene expression profiling[J].Genome Research,2009,19(10):1825-1835.
[7] Elaine R M.Next-generation DNA sequencing methods[J].Annu.Rev.Genomics Hum.Genet.,2008(9):387-402.
[8] Rebecca C,Olivia A,Pamela A H.The gext generation:using new sequencing technologies to analyse gene regulation[J].Respirology,2011,16(2):210-222.
[9] 袁德义,邹锋,谭晓风,等.油茶花芽分化及雌雄配子体发育的研究[J].中南林业科技大学学报,2011,31(3):65-70.
[10] 王湘南,蒋丽娟,陈永忠,等.油茶花芽分化的形态解剖学特征观测[J].中南林业科技大学学报,2011,31(8):22-27.
[11] 罗塔.油茶花芽分化过程形态及内含物质变化的研究[D].广州:华南农业大学,2012.
[12] Michiel J L,Seiya I,John N,etal.Open source clustering Software[J].Bioinformatics,2004,20(9):1453-1454.
[13] Michael B E,Paul T S,Patrick O B,etal.Cluster analysis and display of genome-wide expression patterns[J].Proceedings of the National Academy of Sciences,1998,95(25):14863-14868.
[14] 宁顺斌,王玲,宋运淳.几种差别表达基因显示技术及其在植物方面的应用[J].生命科学,1999,11(3):140-144.
[15] Kim S Y,Randal B,Jerome G,etal.Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection:evidence for differential gene expression.[J].Journal of Virology,1989,63(9):3708-3713.
[16] 胡玉玲,姚小华,任华东,等.油茶花发育转录组测序及相关基因表达分析[J].林业科学,2014,50(9):36-43.
[17] Soraya P,Gary S D,Elvira B,etal.B and C Floral organ identity functions require sepallata mads-box genes[J].Nature,2000,405(6783):200-203.
[18] Detlef W,Elliot M M.The abcs of floral homeotic genes[J].Cell,1994,78(2):203-209.
[19] Heike W,Erica M,Marco T,etal.On reconciling the interactions between apetala2,Mir172 and agamous with the Abc model of flower development[J].Development,2010,137(21):3633-3642.
[20] Enrico C.Goethe and the Abc model of flower development[J].Comptes rendus De L’académie Des Sciences-series Iii-sciences De La Vie,2001,324(6):523-530.
[21] Elena R A,Eugenio A,Rafael B,etal.From Abc genes to regulatory networks,epigenetic landscapes and flower morphogenesis:making biological sense of theoretical approaches[C].Seminars in Cell & Developmental Biology,Elsevier,2010:108-117.
猜你喜欢
黑龙江粮食(2022年6期)2022-11-23
锦绣·上旬刊(2020年10期)2020-12-14
落叶果树(2020年3期)2020-06-17
心电与循环(2020年1期)2020-02-27
河北果树(2020年1期)2020-02-09
中学课程辅导·教师教育(上、下)(2018年15期)2018-10-10
江苏农业科学(2017年5期)2017-04-15
中国马铃薯(2015年3期)2015-12-19
湖北农业科学(2014年3期)2014-07-21
特产研究(2014年4期)2014-04-10
