
The role of DJ-1 complexes and catecholamine metabolism: relevance for familial and idiopathic Parkinson’s disease
2018-05-31 09:02DominikPiston,MatthewE.Gegg
中国神经再生研究(英文版) 2018年5期
Autosomal recessive mutations in thePARK7gene, which encodes for the protein DJ‐1, result in a loss of function and are a cause of familial Parkinson’s disease (PD), while increased wild‐type DJ‐1 protein levels are associated with some forms of cancer. Several functions of DJ‐1 have been described, with the greatest evidence indicating that DJ‐1 is a redox‐sensitive protein involved in the regulation of oxidative stress and cell survival. We have recently reported that the levels of DJ‐1 oxidized at cysteine 106 (C106) was decreased in the cortex of idiopathic PD brains (Piston et al., 2017). Furthermore we found that DJ‐1 forms high molecular weight complexes in human brain and the dopaminergic SH‐SY5Y neuroblastoma cell line, and that these complexes could be oxidized at C106. Proteomics indicated that proteins involved in RNA transcription/translation were associated with these DJ‐1 complexes, and the composition of complexes was affected by oxidation of DJ‐1. RNA sequencing highlighted that transcripts associated with the catecholamine system, including dopamine (DA) metabolism, tended to be increased when complexes contained DJ‐1 mimicking oxidation at C106. DJ‐1 knock down (KD)cells also had increased intracellular DA and noradrenaline (NA)levels. In this perspective we will discuss the implications of DJ‐1 acting as a redox sensor directly affecting RNA metabolism, and with respect to PD, how dysregulation of catecholamine metabolism in both familial and idiopathic PD, might contribute to some prodro‐mal features of the disease and the increased susceptibility of specific neuronal populations to neurodegeneration.
DJ-1 complexes associated with RNA metabolism and affected by oxidation at C106:We have found that DJ‐1 forms high molecular weight complexes in human brain and neuroblastoma cells, with these complexes appearing to contain RNA metabolism proteins such as heterogeneous ribonucleoproteins (hnRNP) and polyadenylate binding protein 1 (PABP1). Association of DJ‐1 with these proteins was affected by the oxidation status of DJ‐1 (Piston et al., 2017). Our finding adds to previous reports of DJ‐1 being directly associated with RNA (Hod et al., 1999; van der Brug et al., 2008) and likely adds another level of transcriptional/translational control to signaling pathways previously reported to be affected by the redox‐status of DJ‐1 such as antioxidant defence and survival/apoptosis (Biosa et al.,2017), in addition to our finding in neuroblastoma cells on catecholamine metabolism.
Several DJ‐1 complexes were detected in human brain lysates and could well reflect the different compositions in neurons and glia,where several different functions of DJ‐1 are described in the two cell types. It is likely that alternate complex composition might also contribute to the differing effects of DJ‐1 described in several types of cancer and cardiomyocytes following ischaemia.
It is becoming increasingly apparent that the oxidation status of C106 is not a simple ‘on‐off’ switch, but rather biphasic. C106 resides in a pocket, and the transition from oxidised C106 (SO2–) to over oxidised C106 (e.g., SO3–) has been proposed to change the local con‐formation of the protein resulting in DJ‐1 becoming destabilized and losing function (Cao et al., 2014; Kiss et al., 2017). Therefore it can be envisaged that the composition of DJ‐1 complexes under acute or mild oxidative stress will differ from that of chronic excessive oxidizing conditions, thus altering the physiological response of the cell over time, for example from pro‐survival to apoptotic (Cao et al.,2014).
The observation that DJ‐1 complexes are oxidized in idiopathic PD brains, and that DJ‐1 oxidation appears to be decreased in frontal cortex of PD brains, raises the prospect that changes in RNA metabolism also occur in idiopathic PD. Oxidised DJ‐1 at C106 by immunohistochemistry has also been reported in neurons and glia of PD brains, and seems to diminish with disease progression (Saito et al., 2014). DJ‐1 has been proposed as a biomarker for PD and certain types of cancer such as breast and lung. Given the apparent import‐ant role of C106 oxidation in controlling DJ‐1 function, in future,measuring the ratio of oxidized to reduced DJ‐1 in cerebro‐spinal fluid (CSF)/serum might prove more informative.
DJ-1 and the catecholamine system:In terms of PD, the finding in neuroblastoma cells expressing an oxidation mimic of C106 that several transcripts associated with catecholamine metabolism were affected fits well with both the motor and non‐motor symptoms of the disease. Upregulated transcripts included dopa decarboxylase(DDC) involved in DA synthesis, vesicular monoamine transporter 1 (VMAT1), required for the sequestration of DA into vesicles; and dopamine beta hydroxylase (DBH) which converts DA to NA in vesicles (Piston et al., 2017). Furthermore in DJ‐1 KD cells, in which no DJ‐1 complexes were detectable, and presumably resulting in the loss of transcriptional/translational control of transcripts, both the levels of DA and NA were significantly increased (Piston et al., 2017).DJ‐1 function has previously been associated with DA metabolism,including both transcriptional control of tyrosine hydroxylase (TH),the first step in DA synthesis, and direct binding to and activation of TH and DDC by DJ‐1. DJ‐1 knockout mice also exhibit increased DA reuptake, linked to increased activity of the DA transporter DAT,and perhaps the inhibition of DA D2 receptor, involved in the control of DA synthesis and release. Further work is required to establish whether the elevation in these two neurotransmitters in DJ‐1 KD cells is due to increased synthesis, decreased turnover, and/or accu‐mulation of these molecules in the cytoplasm, rather than vesicles.
Dysregulated catecholamine metabolism in familial and idiopathic PD:Both DA and NA are readily oxidized, forming toxic intermediates that are implicated in neurodegeneration. It is notable that two brain regions with considerable neuronal loss in PD are the DA neurons of the substantia nigra pars compacta and NA neurons of the locus coeruleus. Both neuronal populations contain the pigment neuromelanin, which contains oxidation products of DA and NA,and is formed when these neurotransmitters are not sequestered in vesicles. Furthermore, many of the non‐motor functions associated with PD including prodromal features such as constipation, sleep disruption and depression, and later features such as cognitive impairment can all be linked to DA and/or NA systems (Schapira et al.,2017) (Figure 1). It has been proposed that the broad action potentials and autonomous pacemaking, which results in large oscillations of cytosolic calcium, contribute to the selective vulnerability of DA neurons in the substantia nigra pars compacta. Recently Burbulla et al. (2017) have shown that levels of oxidised DA in the cytosol increase over time in midbrain DA neurons with DJ‐1 KO or DJ‐1 mutations. They propose that increased oxidative stress as a result of dysfunctional DJ‐1 and high calcium levels is responsible. Intriguingly, one consequence of this is inhibition of the lysosomal enzyme glu‐cocerebrosidase (GCase). Mutations in theGBAgene, which encodes GCase, cause 5–10% of all PD cases. Furthermore, GCase activity is decreased in idiopathic PD brains, with the substantia nigra showing the greatest deficit (Gegg et al., 2012). In light of our findings, we pro‐pose that dysregulation of catecholamine homeostasis, and in particular increased DA levels, might also contribute to this phenomenon(Figure 1). Indeed, Burbulla et al indicate that increasing DA levels in mouse neurons with L‐DOPA elevated DA oxidation and inhibition of GCase. Oxidised DA was also increased but to a lesser extent in neurons withPINK1andparkinmutations, and GCase activity is diminished in dopaminergic cells lacking these functional proteins(Gegg et al., 2012; Burbulla et al., 2017). Mutations in both proteins are also associated with increased oxidative stress and mitochondrial dysfunction and likely contribute to the phenomena. DJ‐1,PINK1andparkinmouse models all exhibit perturbed striatal DA function,that precedes nigral‐striatal degeneration, once again implicating dysregulated catecholamine metabolism (Kitada et al., 2009).
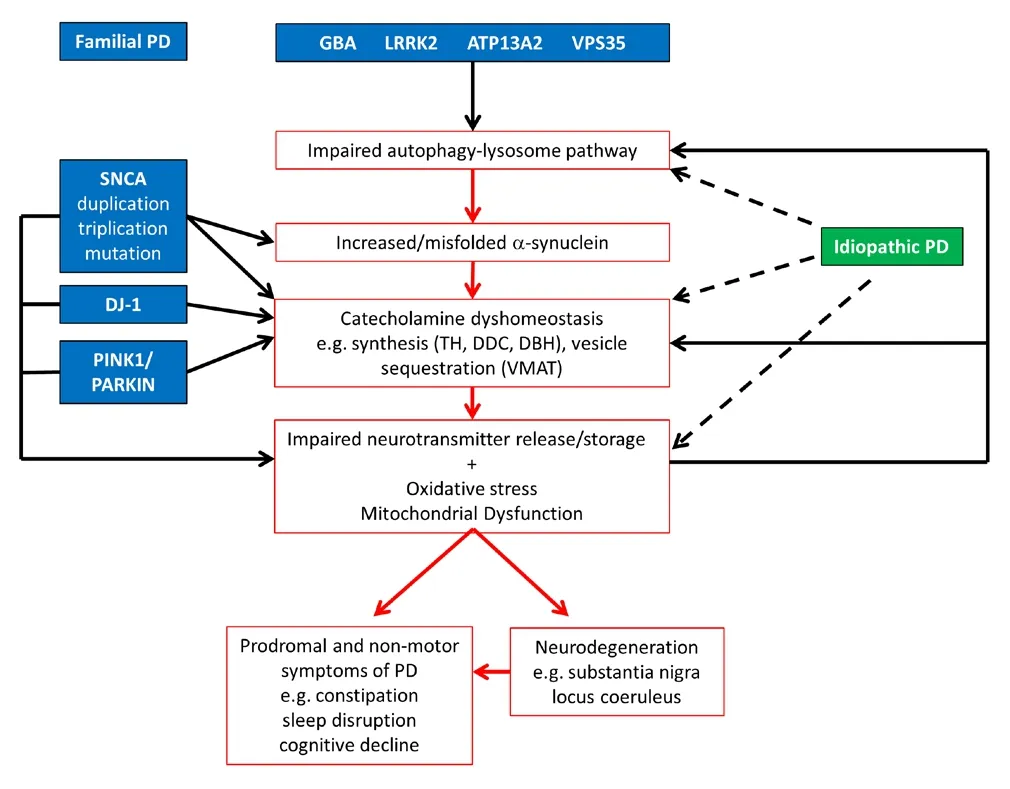
Figure 1 Pathogenic pathways of idiopathic and familial forms of Parkinson’s disease (PD).
Can other genetic causes of PD or the idiopathic forms of the disease also be linked to catecholamine dyshomeostasis?The accumulation of α‐synuclein in Lewy bodies is a hallmark of PD. While the exact function(s) of α‐synuclein is still unclear, the protein is known to predominantly localize to presynaptic terminals and bind highly curved membranes, including neurotransmitter vesicles. α‐synuclein has been proposed to be involved in neurotransmitter release, but in a regulatory, rather than an essential role, as it is generally lacking in inhibitory neurons, is one of the last proteins to reach the synapse and is absent in invertebrates (Burré, 2015). α‐Synuclein has been proposed to play roles in vesicle filling, clustering, soluble N‐ethyl‐maleimide‐sensitive factor attachment protein receptor (SNARE)complex formation and the synaptic vesicle cycle (Burré, 2015). In‐creased levels of α‐synuclein, as a result of duplication or triplication of the α‐synuclein gene, or aberrant membrane binding due to point mutations, could all disrupt the loading/release of catecholamines and increase toxic oxidized species. Lysosomal function is known to decline with age, the greatest risk factor for PD, and inhibition of the autophagy‐lysosomal pathway causes accumulation and/or oligo‐merization/fibrillation of α‐synuclein. Mutations inGBA,LRRK2,VPS35andATP13A2are all known to inhibit autophagy, resulting in accumulation of α‐synuclein. Lrrk2 has also been linked with rab proteins, which are important for vesicle trafficking. Midbrain DA neurons containingLrrk2mutations or triplication of the α‐synuclein gene exhibit increased levels of oxidised DA (Burbulla et al., 2017).Therefore in addition to the mitochondrial dysfunction and oxidative stress discussed above, impaired lysosomal function and impaired vesicle trafficking could also contribute to dysregulated intracellular catecholamine levels and neurodegeneration, and likely feedback on one another in both genetic and idiopathic PD (Figure 1).
Conclusion:Measuring the levels of monomeric and oxidized DJ‐1 described above, as well as DA, NA and their metabolites in cell mod‐els and CSF of PD patients could provide an insight to disease mechanisms. For example the ratio of DA to its precursor DOPA or oxidized forms such as 3,4‐dihydroxyphenylacetic acid (DOPAC), which results from the oxidation of cytosolic DA in neurons by monoamine oxidase A, might indicate the points at which DA/NA metabolism are affected (e.g., synthesis or cytosolic accumulation of neurotrans‐mitters). Furthermore, given the involvement of the catecholamine system in several prodromal features of PD, measurement of these metabolites in the CSF from at risk individuals such as mutantGBAcarriers could help identify useful biomarkers for disease progression.
Due to the restriction on both the number of words and references we would like to thank the authors of the papers we were unable to cite in this perspective.
This work was funded by a Medical Research Council (UK) Experimental Medicine grant [MR/M006646/1].
Dominik Piston, Matthew E. Gegg*
German Centre for Neurodegenerative Diseases (DZNE), Bonn,Germany (Piston D)
Department of Clinical Neuroscience, UCL Institute of Neurology,London, UK (Gegg ME)
*Correspondence to:Matthew E. Gegg, Ph.D.,matthew.gegg@ucl.ac.uk.
orcid:0000-0001-8093-0723 (Matthew E. Gegg)
Accepted:2018-03-17
doi:10.4103/1673-5374.232474
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer review report:
Reviewer:Chih-Li Lin, Chung Shan Medical University, China.
Comments to authors:Authors reported a perspective review discussing the neuroprotective function of DJ-1 in PD. According to their recent relevant publication, authors provided an insight for the role of DJ-1 in PD pathogenesis, particularly for that the oxidative changes to DJ-1 are concomitant with changes in mRNA transcripts and involved in catecholamine metabolism.This is a well-written short manuscript that deals with molecular mechanisms of microglial regulation in the development of brain.
Biosa A, Sandrelli F, Beltramini M, Greggio E, Bubacco L, Bisaglia M (2017)Recent findings on the physiological function of DJ‐1: Beyond Parkin‐son’s disease. Neurobiol Dis 108:65‐72.
Burbulla LF, Song P, Mazzulli JR (2017) Dopamine oxidation mediates mi‐tochondrial and lysosomal dysfunction in Parkinson’s disease. 357:1255‐1261.
Burré J (2015) The synaptic function of alpha‐synuclein. J Parkinsons Dis 5:699‐713.
Cao J, Ying M, Xie N, Lin G, Dong R, Zhang J, Yan H, Yang X, He Q, Yang B (2014) The oxidation states of DJ‐1 dictate the cell fate in response to oxidative stress triggered by 4‐hpr: autophagy or apoptosis? Antioxid Redox Signal 21:1443‐1459.
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH (2012) Glucocerebrosidase deficiency in substantia nigra of parkin‐son disease brains. Ann Neurol 72:455‐463.
Hod Y, Pentyala SN, Whyard TC, El‐Maghrabi MR (1999) Identification and characterization of a novel protein that regulates RNA‐protein inter‐action. J Cell Biochem 72:435‐444.
Kiss R, Zhu M, Jójárt B, Czajlik A, Solti K, Fórizs B, Nagy É, Zsila F,Beke‐Somfai T, Tóth G (2017) Structural features of human DJ‐1 in distinct Cys106 oxidative states and their relevance to its loss of function in disease. Biochim Biophys Acta 1861:2619‐2629.
Kitada T, Tong Y, Gautier CA, Shen J (2009) Absence of nigral degeneration in aged parkin/DJ‐1/PINK1 triple knockout mice. J Neurochem 111:696‐702.
Piston D, Alvarez‐Erviti L, Bansal V, Gargano D, Yao Z, Szabadkai G, Odell M, Puno MR, Bjorkblom B, Maple‐Grodem J, Breuer P, Kaut O, Larsen JP, Bonn S, Moller SG, Wullner U, Schapira AHV, Gegg ME (2017) DJ‐1 is a redox sensitive adapter protein for high molecular weight complexes involved in regulation of catecholamine homeostasis. Hum Mol Genet 26:4028‐4041.
Saito Y, Miyasaka T, Hatsuta H, Takahashi‐Niki K, Hayashi K, Mita Y, Ku‐sano‐Arai O, Iwanari H, Ariga H, Hamakubo T, Yoshida Y, Niki E, Mu‐rayama S, Ihara Y, Noguchi N (2014) Immunostaining of oxidized DJ‐1 in human and mouse brains. J Neuropathol Exp Neurol 73:714‐728.
Schapira AHV, Chaudhuri KR, Jenner P (2017) Non‐motor features of Par‐kinson disease. Nat Rev Neurosci 18:435‐450.
van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan‐Mamczarz K, Martindale J, Xie C, Ahmad R, Thomas KJ, Beilina A,Gibbs JR, Ding J, Myers AJ, Zhan M, Cai H, Bonini NM, Gorospe M,Cookson MR (2008) RNA binding activity of the recessive parkinsonism protein DJ‐1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci U S A 105:10244‐10249.
- 中国神经再生研究(英文版)的其它文章
- Novel function of the chemorepellent draxin as a regulator for hippocampal neurogenesis
- Weak phonation due to unknown injury of the corticobulbar tract in a patient with mild traumatic brain injury: a diffusion tensor tractography study
- Semaphorin 3A: from growth cone repellent to promoter of neuronal regeneration
- The role of undifferentiated adipose-derived stem cells in peripheral nerve repair
- Nerve conduction models in myelinated and unmyelinated nerves based on three-dimensional electrostatic interaction
- Fatigability during volitional walking in incomplete spinal cord injury: cardiorespiratory and motor performance considerations