
Magnesium acethyltaurate as a potential agent for retinal and optic nerve protection in glaucoma
2018-05-31 09:02:26IgorIezhitsa,RenuAgarwal
中国神经再生研究(英文版) 2018年5期
Glaucoma is the second leading cause of irreversible vision impairment affecting more than 70 million people worldwide with approximately 10%suffering from glaucoma‐related bilateral blind (Quigley and Broman,2006). It is a multi‐factorial disease that is characterized by optic nerve damage and visual field loss. Progressive loss of retinal ganglion cells(RGCs) resulting in visual field deficits is the hallmark of glaucoma. Sever‐al etiological factors seem to be involved in its pathophysiology, however the precise mechanisms leading to its development remain unclear. Some of the widely described factors that contribute to RGC loss include axonal transport failure, neurotrophic factor depletion, excitotoxicity, mitochon‐drial dysfunction, activation of intrinsic and extrinsic apoptotic cascade and oxidative stress (Agarwal et al., 2009). The basic mechanisms that contribute to RGC degeneration in glaucoma are presented in Figure 1.Current treatment strategies for glaucoma are limited to the reduction of intraocular pressure (IOP); however, it is clear now that the disease pro‐gression may continue despite effective IOP lowering. Search for newer modalities has led to emergence of significant data from experimental re‐search suggesting potentially new neuroprotective strategies that may add to the arsenal of existing antiglaucoma agents (Vasudevan et al., 2011).
One of the possible targets for neuroprotection could be the endo‐thelin (ET) and N‐methyl‐D‐aspartate (NMDA) mediated signaling in retina. Elevated ET‐1 levels as well as excessive glutamate mediated neurotransmission have been observed in glaucomatous eyes. ET‐1, a potent vasoconstrictor, causes retinal ischemia that potentiates gluta‐mate mediated excitotoxicity. ET‐1, in fact, may act synergistically with glutamate to damage retinal neurons. Glutamate‐induced neurotoxicity involves NMDA receptor activation leading to in flux of Ca2+and ET‐1 is also known to increase the intracellular Ca2+concentration in glial and neuronal cells. Additionally, ET‐1 specifically stimulates the efflux of glutamateviaET receptors from astrocytes, suggesting that ET‐1 exacerbates neurodegeneration (Kobayashi et al., 2005). Hence, several studies have been done to investigate therapies for RGC protection by targeting ET‐1 and NMDA signaling. Accordingly, NMDA and/or ET receptor antagonists have been suggested to inhibit excitotoxic RGC loss and to delay progression of visual loss in glaucoma. In a series of our recent studies we examined the potential of neuroprotective effect of magnesium (Mg)acetyltaurate (MgAT) against retinal and optic nerve damage induced by NMDA and ET‐1. The choice of MgAT was due to structural combination of Mg and taurine as both were expected to provide neuroprotection through multiple mechanisms such as inhibition of ET‐1 effect, antagonism of NMDA receptors, and antioxidant effects.
We observed that exposure to both the NMDA and ET‐1 results in significant increase in the number of TdT‐mediated dUTP nick‐end la‐beling (TUNEL)‐positive (apoptotic) cells on day 7 post‐exposure in rat retinas. However, the number of apoptotic retinal cells was significantly reduced in rats treated with MgAT, particularly in rats that received MgAT as pre‐treatment. Consistent with the findings of TUNEL staining,hematoxylin and eosin stained retinal sections showed that the number of neuronal cells in ganglion cell layer (GCL) was strikingly reduced in retinas 7 days after both NMDA and ET‐1 exposure. By contrast, retinal cell number was preserved in inner retina of animals pretreated with MgAT. We also examined the optic nerve morphology among groups of rats treated with NMDA or ET‐1 with and without MgAT using toluidine blue stained optic nerve sections. Clearly NMDA and ET‐1 induced ax‐onal loss was attenuated by treatment with MgAT, and in particular the pre‐treatment. Overall, these data demonstrated that the pre‐treatment with MgAT was more effective than co‐ and post‐treatment in preventing NMDA and ET‐1 induced retinal and optic nerve damage (Arfuzir et al.,2016; Jafri et al., 2017; Lambuk et al., 2017).
We also observed that intravitreal NMDA injection causes an increase in Ca, Mg, Na and K contents of retinas with significant (by 3 and 1.27 times) increase of Ca/Mg and Na/K ratios compared to control group.Glutamatergic excitotoxicity, originally described in the retina, primarily results from intracellular accumulation of calcium ions. Because of the relatively high permeability of NMDA‐type of glutamate‐gated channels to Ca2+ions, neurons are particularly sensitive to injury associated with excessive activity of this channel subtype. Both decreased and elevated levels of minerals can cause RGC dysfunction. Pre‐treatment with MgAT abolished the effect of NMDA and was more effective that co‐ and post‐treatment. The trace elements such as Cu, Fe and Zn also showed a significant increase compared to control group in NMDA treated rat retinas. Treatment with MgAT abolished the effect of NMDA. Overall,pretreatment with MgAT was more effective than co‐treatment with MgAT in restoring both the minerals and trace element contents of retinas (Jafri et al., 2017).
Oxidative stress as an important contributor to glutamate‐induced RGCs loss has been described widely. Consequently, enhancement of the antioxidant defenses of the retina could prevent or delay the RGC loss.To evaluate the effect of MgAT on oxidative stress, we quantified retinal reduced glutathione (GSH), superoxide dismutase (SOD), catalase (CAT)and malondialdehyde (MDA). We observed that intravitreal NMDA as well as ET‐1 exposure causes significant decrease in the levels of CAT and SOD activity and GSH contents and a significant increase in the levels of MDA indicating oxidative stress as a result of overproduction of free radicals and lipid peroxide and/or a significantly reduced antioxidant defenses (Arfuzir et al., 2016; Jafri et al., 2017; Lambuk et al., 2017). Rats treated with MgAT showed significant improvement in all parameters of oxidative stress and this improvement was particularly evident in the pretreatment group rather than in co‐ and post‐treatment groups, thus further confirming the role of MgAT in protecting retina from NMDA or ET‐1 induced oxidative stress (Arfuzir et al., 2016; Jafri et al., 2017; Lam‐buk et al., 2017). It has been reported that SOD and catalase play an important role as the first line defense against reactive oxygen species (ROS)mediated changes. Activity of catalase is secondary to SOD and thus limits the accumulation of H2O2, which is generated by various oxidases in tissue. Magnesium is an essential cofactor for GSH synthesis and in all biosynthetic reactions involving ATP (Zheltova et al., 2016). Maintenance of GSH in its reduced form is important to prevent the protein‐thi‐ol mixed disulfide formation. In our study, MgAT treatment significantly increased retinal GSH levels compared to vehicle treated group.
In our study, MDA concentration in NMDA‐ and ET‐1‐treated groups were significantly higher than in the other groups, indicating increased lipid peroxidation. However, in all MgAT groups, the MDA concentration was comparable to control group. The elevated level of MDA in retina may be due to the poor retinal antioxidant capacity as a result of low GSH levels. Thus, from our results it seemed that MgAT increases antioxidant activity leading to inhibition of lipid peroxidation that is reflected by reduced MDA level in retina (Arfuzir et al., 2016; Jafri et al.,2017; Lambuk et al., 2017).
Brain derived neurotrophic factor (BDNF) is a widely expressed neurotrophin in retinal tissue. It is crucial for normal neuronal development and maintenance and, with enhanced expression, offers protection against injurious stimuli. In our study BDNF level was significantly depleted in retina on day 7 after NMDA exposure while treatment with MgAT caused BDNF upregulation with restoration of retinal and optic nerve morphology (Lambuk et al., 2017). Among various neurotrophic factors, BDNF in particular has been shown to prevent glutamate‐me‐diated cell death in several neuronal populations bothin vitroandin vivo. Rather, RGC survival may depend on a sufficient level of BDNF and its receptor expression in NMDA‐damaged cells. Besides affecting expression of BDNF and its receptors, excitotoxicity may also interrupt retrograde transport in the optic nerve, hence depriving neuronal cell bodies of molecules such as BDNF that are required for the survival of RGCs. While this alteration in retrograde transport is present, BDNF protein synthesis in RGCs is stimulated, at least during the first few hours after damage, corroborating the idea of locally producedvs. retrogradely transported BDNF. This increased synthesis of BDNF may represent an endogenous neuroprotective response by RGCs (Vecino et al., 2002).However, after this initial response there is a decrease in the expression of BDNF in RGCs which could be due to an alteration in the metabolism of the affected cells or also to an interruption of BDNF retrograde transport(Vecino et al., 2002).
The final common pathway for neuronal cell death is necrosis or apop‐tosis, the latter playing a major role in RGC death in glaucoma. RGC death in response to various stimuli may involve a variety of molecular pathways, both regulatory and executioner. Regardless of the initiating injury, apoptotic loss of RGCs involves activation of the caspase cascade,increased expression of pro‐apoptotic genes such as Bax/Bid and down‐regulation of anti‐apoptotic genes such as Bcl‐2/Bcl‐xl. NMDA exposure increases the ratio of the pro‐apoptotic protein Bax to the anti‐apoptotic protein Bcl‐2 by directly cleaving Bax to an activated form through a mi‐tochondrial pathway (Wood and Newcomb, 2000). Activated Bax forms multimers in the mitochondria, with subsequent cytochrome c release,which ultimately contributes to apoptosisviaformation of the apopto‐some protein complex and activation of downstream caspases 3 and 7.In our studies, exposure to both NMDA and ET‐1, caused activation of retinal caspase‐3. Remarkably there was a 50 fold increase in the Bax/Bcl‐2 ratio in NMDA exposed retina providing further evidence that NMDA as well as ET‐1 exposures causes disruption of this ratio leading to RGC apoptosis. Moreover, pretreatment with MgAT reduces caspase‐3 activation and restores the Bax/Bcl‐2 ratio to control levels in NMDA‐exposed retinas, indicating that treatment with MgAT can attenuate pro‐apoptotic sequences perhaps at the mitochondrial level (Lambuk et al., 2017).
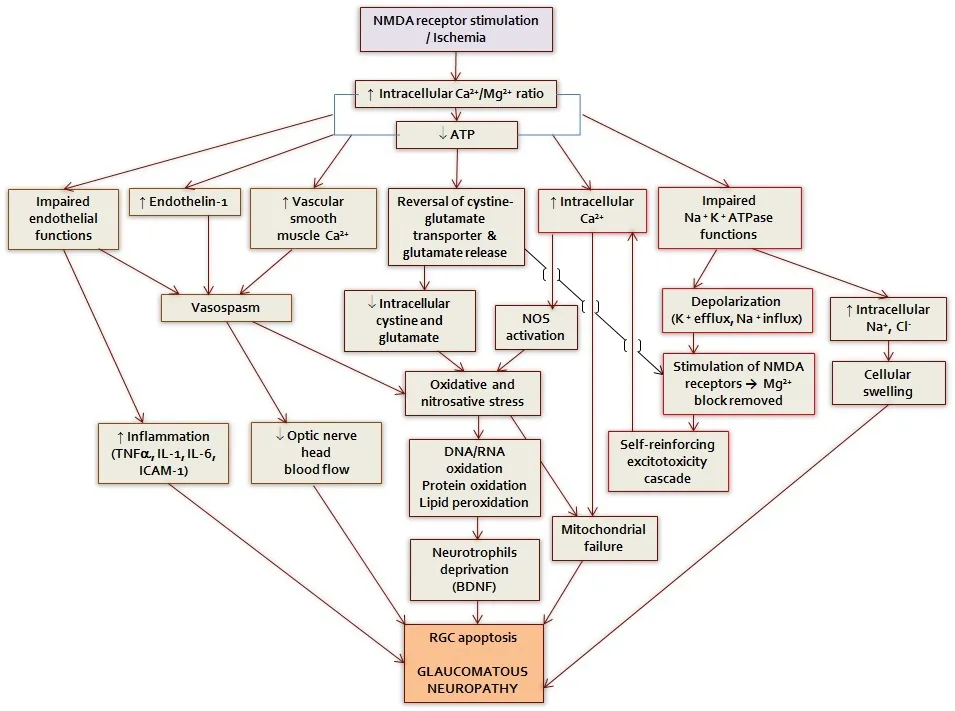
Figure 1 The basic mechanisms that contribute to retinal ganglion cell degeneration in glaucoma.
In line with the observations stated here, we have previously described the role of Mg in ophthalmic diseases (Agarwal et al., 2013, 2014). The neuroprotective effects of taurine alone are also well established. Retinal taurine level is crucial in preventing RGC damage in major retinal diseases. It was found that taurine can directly prevent RGC degeneration, oc‐curring either in serum‐deprived pure RGC cultures or in animal models representing RGC loss. Furthermore, taurine is known to partly prevent NMDA‐induced RGC excitotoxicity. Hence, MgAT, a combined salt of Mg and taurine, seems to be effective in preventing NMDA and ET‐1 in‐duced retinal and optic nerve damage by counteracting NMDA receptor activation, reducing retinal oxidative stress, increasing retinal BDNF level and reducing activation of caspase and pro‐apoptotic proteins
Based on the investigations done so far, it can be concluded that MgAT has protective effects against NMDA and ET‐1 induced retinal and optic nerve damage. Considering the enhanced efficacy of MgAT particularly on pretreatment, it is likely that MgAT primarily has therapeutic potential as a protective agent in high risk individuals. Development of appropriate formulations to administer MgAT noninvasively would be the key to its clinical application. Furthermore, investigations into the mechanisms of action of MgAT will reveal its true potential as an antiglaucoma agent.
The work was supported by Institut Pengurusan Penyelidikan (RMI),Universiti Teknologi MARA, Malaysia, under the grant No. 600-IRMI/MyRA 5/3/LESTARI (0088/2016) and 600-IRMI/DANA 5/3/LESTARI(0076/2016).
Igor Iezhitsa*, Renu Agarwal
Centre for Neuroscience Research, Faculty of Medicine, Universiti Teknologi MARA, Selangor, Malaysia (Iezhitsa I, Agarwal R)Volgograd State Medical University, Research Institute of Pharmacology,Volgograd, Russia (Iezhitsa I)
*Correspondence to:Igor Iezhitsa, Dr. Sci. Biol., Ph.D.,iezhitsa@salam.uitm.edu.my or iezhitsa@yandex.ru.
orcid:0000-0002-2852-8486 (Igor Iezhitsa)
Accepted:2018-03-19
doi:10.4103/1673-5374.232470
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Georgios D Panos, Geneva University Hospitals, Switzerland.
Agarwal R, Iezhitsa L, Agarwal P (2014) Pathogenetic role of magnesium deficiency in ophthalmic diseases. Biometals 27:5‐18.
Agarwal R, Iezhitsa IN, Agarwal P, Spasov AA (2013) Mechanisms of cataractogenesis in the presence of magnesium deficiency. Magnes Res 26:2‐8.
Agarwal R, Gupta SK, Agarwal P, Saxena R, Agrawal SS (2009) Current concepts in the pathophysiology of glaucoma. Indian J Ophthalmol 57:257‐266.
Arfuzir NN, Lambuk L, Jafri AJ, Agarwal R, Iezhitsa I, Sidek S, Agarwal P, Bakar NS,Kutty MK, Yusof AP, Krasilnikova A, Spasov A, Ozerov A, Mohd Ismail N (2016)Protective effect of magnesium acetyltaurate against endothelin‐induced retinal and optic nerve injury. Neuroscience 325:153‐164.
Jafri AJA, Arfuzir NNN, Lambuk L, Iezhitsa I, Agarwal R, Agarwal P, Razali N,Krasilnikova A, Kharitonova M, Demidov V, Serebryansky E, Skalny A, Spasov A,Yusof APM, Ismail NM (2017) Protective effect of magnesium acetyltaurate against NMDA‐induced retinal damage involves restoration of minerals and trace elements homeostasis. J Trace Elem Med Biol 39:147‐154.
Kobayashi T, Oku H, Fukuhara M, Kojima S, Komori A, Ichikawa M, Katsumura K,Kobayashi M, Sugiyama T, Ikeda T (2005) Endothelin‐1 enhances glutamate‐in‐duced retinal cell death, possibly through ETA receptors. Invest Ophthalmol Vis Sci 46:4684‐4690.
Lambuk L, Jafri AJ, Arfuzir NN, Iezhitsa I, Agarwal R, Rozali KN, Agarwal P, Bakar NS, Kutty MK, Yusof AP, Krasilnikova A, Spasov A, Ozerov A, Ismail NM (2017)Neuroprotective effect of magnesium acetyltaurate against nmda‐induced excito‐toxicity in rat retina. Neurotox Res 31:31‐45.
Quigley HA, Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90:262‐267.
Vasudevan SK, Gupta V, Crowston JG (2011) Neuroprotection in glaucoma. Indian J Ophthalmol 59 Suppl:S102‐113.
Vecino E, García‐Crespo D, García M, Martinez‐Millán L, Sharma SC, Carrascal E (2002) Rat retinal ganglion cells co‐express brain derived neurotrophic factor(BDNF) and its receptor TrkB. Vision Res 42:151‐157.
Wood DE, Newcomb EW (2000) Cleavage of Bax enhances its cell death function. Exp Cell Res 256:375‐382.
Zheltova AA, Kharitonova MV, Iezhitsa IN, Spasov AA (2016) Magnesium deficiency and oxidative stress: an update. Biomedicine (Taipei) 6:8‐14.
- 中国神经再生研究(英文版)的其它文章
- Weak phonation due to unknown injury of the corticobulbar tract in a patient with mild traumatic brain injury: a diffusion tensor tractography study
- Exosomes: a novel therapeutic target for Alzheimer’s disease?
- Inhibition of retinal ganglion cell apoptosis:regulation of mitochondrial function by PACAP
- Trillium tschonoskii maxim extract attenuates abnormal Tau phosphorylation
- Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction
- Nogo receptor expression in microglia/macrophages during experimental autoimmune encephalomyelitis progression