
齐墩果酸糖苷衍生物的合成及抗肿瘤活性的研究
2018-05-30 10:36鹿学宇孟艳秋
沈阳化工大学学报 2018年1期
汤 义, 鹿学宇, 孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
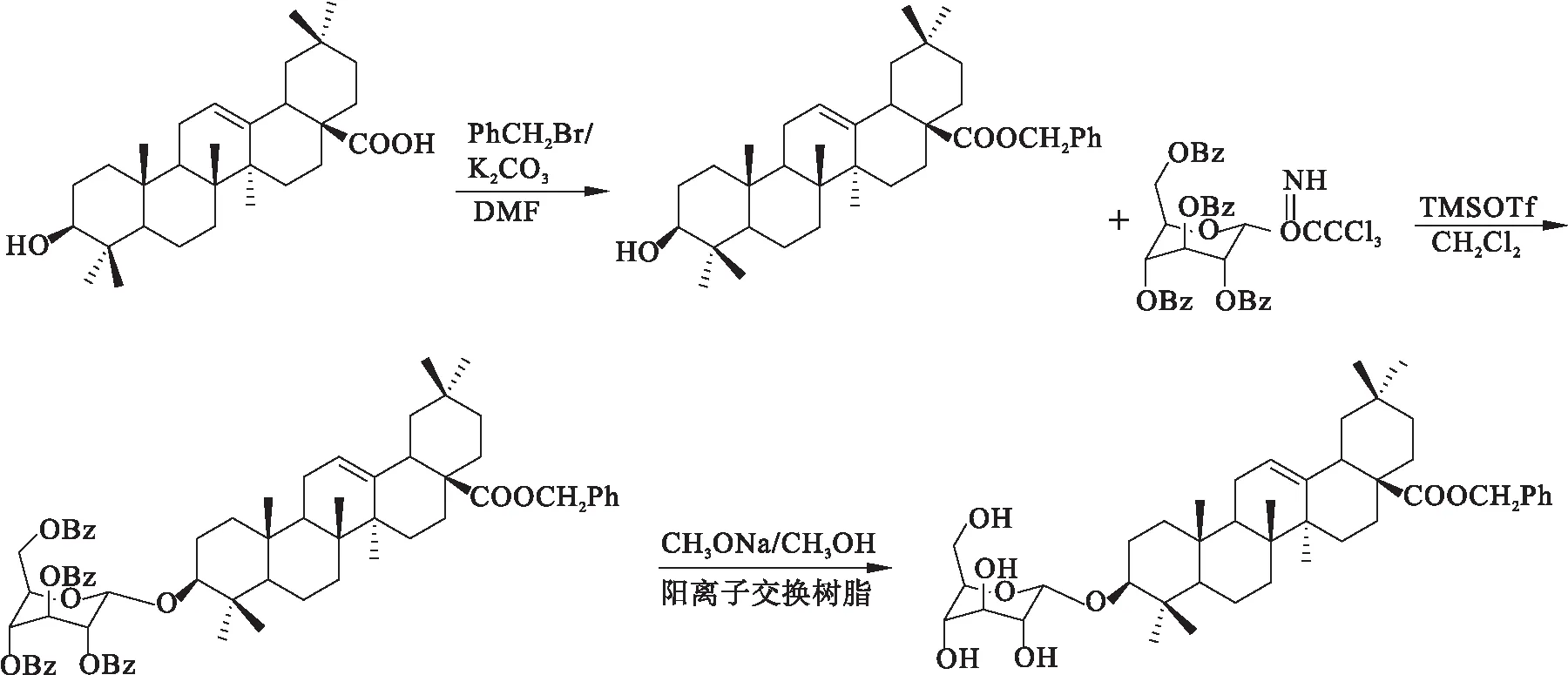
齐墩果酸(Oleanic acid,OA)又名庆四素,化学名3β-羟基-齐墩果烷型-12-烯-28-酸,属于五环三萜类化合物,多以皂苷形式存在,具有广泛的生物学效应,如保肝、抗炎、抗肿瘤、抗艾滋病毒等药理作用[1-5].研究表明,C-28位羧基成酯或酰胺的齐墩果酸衍生物具有较强的抗肿瘤活性[6-7],且已有大量研究表明,糖链对三萜类化合物的生物活性关系密切,糖链的改变可能对皂苷的活性产生较大影响,MATSUDA[8]等在研究齐墩果酸皂苷对口服葡萄糖大鼠的血糖水平影响时,发现齐墩果酸3-糖链和28-羧基糖链两个结构对降血糖活性极其重要,并且C-3位连接葡萄糖和葡萄糖醛酸对活性影响显著不同,为了深入研究齐墩果酸结构与其抗肿瘤活性的关系,本文以OA为先导化合物,保留五环三萜骨架,对C-3、C-28位进行结构修饰,合成4个新型齐墩果酸衍生物.以天然皂苷中常见的单糖为原料,经苯甲酰基保护,异头碳溴代,水解,与三氯乙腈成酯等反应合成4个糖苷供体A1~A4[9-10].同时以齐墩果酸为原料,与溴苄反应生成28位苄酯衍生物OA0,在TMSOTf催化下OA0和A1~A4反应生成齐墩果酸糖苷化合物,最后在碱性条件下脱除保护基团得到4个目标产物Ⅰa~Ⅰd.
合成路线如下:
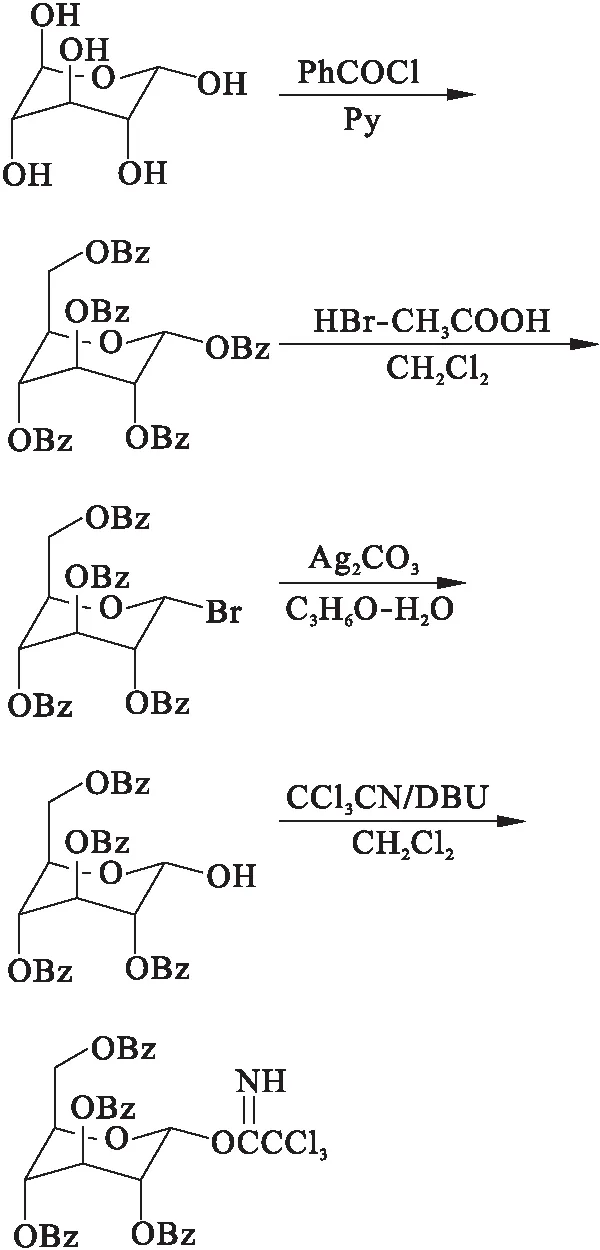
1 实验部分
1.1 主要试剂与仪器
D-葡萄糖,D-木糖,D-半乳糖,L-鼠李糖,国药集团化学试剂有限公司;吡啶,上海实意化学试剂有限公司;苯甲酰氯,上海同方精细化工有限公司;氮杂二环(DBU),Acros;三氟甲磺酸三甲基硅酯(TMSOTf);薄层色谱硅胶GF254,柱色谱硅胶(100~200目,青岛海洋化工厂);齐墩果酸购于陕西森弗生物技术有限公司;Büchi B-540熔点测定仪(未校正);BrukerARX-600型核磁共振分析仪,CDCl3为溶剂,TMS为内标,其他试剂均为分析纯.
1.2 中间体的合成
1.2.1 2,3,4,6-四-O-苯甲酰基葡萄糖三氯乙酰亚胺酯
1.2.1.1 全苯甲酰化葡萄糖的制备
称取1.0 g(5.6 mmol)葡萄糖溶于20 mL无水吡啶,冰浴下缓慢滴加苯甲酰氯5 mL(43.6 mmol),滴加时溶液变浑浊,10 min内滴加完毕.升至室温,反应8 h,向反应液中加甲醇2 mL淬灭反应.减压蒸除溶剂,以二氯甲烷溶解剩余物,分别以稀盐酸30 mL,碳酸氢钠30 mL,饱和食盐水30 mL洗涤3次,分离有机相,无水MgSO4干燥,无需纯化直接进行下一步.收率95 %.mp:178~180 ℃.1H-NMR(600 MHz,CDCl3),δ:8.18~7.30(25H,m,Ar—H);6.36(1H,d,7.8 Hz,H-1);6.10(1H,t,J=7.8 Hz,H-3);5.87(2H,m,H-4,2);4.68,4.52,4.46(3H,m,H-5,6,6′).ESI-MS(m/z):700.21[M+H]+.
1.2.1.2 端基的溴代反应
取3.5 g(5.0 mmol)1.2.1.1的反应产物,溶于20 mL无水二氯甲烷中,加入质量分数33 %氢溴酸醋酸溶液1.5 mL,溶液变成浅黄色,室温搅拌8 h后在冰浴下加入饱和碳酸氢钠溶液20 mL,分离有机相,有机相用饱和食盐水洗涤,硫酸镁干燥,无需纯化直接进行下一步反应.收率87 %.mp:128~130 ℃.1H-NMR(600 MHz,CDCl3),δ:8.10~7.35(20H,m,Ar—H);6.55(1H,d,J=5.6 Hz,H-1);6.25(1H,dd,J=7.9 Hz,5.6 Hz,H-2);5.85(1H,dd,J=7.9 Hz,4.9 Hz,H-4);5.35(1H,t,J=7.9 Hz,H-3);4.85,5.05,5.25(3H,m,6′,6,5).ESI-MS(m/z):659.50[M+H]+.
1.2.1.3 异头碳水解
取2.5 g(4.1 mmol)干燥的溴代苯甲酰基葡萄糖溶于20 mL丙酮中,再加入0.5 mL蒸馏水,加入0.8 g Ag2CO3室温搅拌4 h.加硅藻土抽滤,减压蒸出溶剂丙酮,滴加几滴甲苯将水蒸出,红外灯下干燥.收率80 %.mp:58~63 ℃.1H-NMR(600 MHz,CDCl3),δ:8.05~7.30(20H,m,Ar—H);6.05(1H,d,J=7.7 Hz,H-1);5.64(1H,dd,J=10.2 Hz,7.7 Hz,H-2);5.53(1H,t,J=7.8 Hz,H-3);5.20(1H,dd,J=10.3 Hz,7.2 Hz,H-4);4.62,4.48,4.42(3H,m,5,6,6′).ESI-MS(m/z):596.23[M+H]+.
1.2.1.4 2,3,4,6-四-O-苯甲酰基葡萄糖三氯乙酰亚胺酯
取2.0 g(3.3 mmol)1.2.1.3产物1-羟基苯甲酰葡萄糖溶于30 mL无水二氯甲烷,加入DBU 0.48 mL,搅拌10 min,加入0.6 mL三氯乙腈,搅拌5 h,蒸干溶剂,剩余物溶解于适量二氯甲烷中,以碱化(三乙胺)的中性氧化铝拌样,自然挥干溶剂.用20倍量碱化的中性氧化铝装柱.洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得终产物2.86 g,产率70 %.mp:71~74 ℃.1H-NMR(600 MHz,CDCl3),δ:8.63(1H,s,NH);8.08~7.30(20H,m,Ar—H);6.82(1H,d,J=3.6 Hz,H-1);6.26(1H,t,J=10.2 Hz,H-3);5.82(1H,t,J=10.2 Hz,H-4);4.69(1H,dd,J=3.6 Hz,10.2 Hz,H-2);4.65,4.64,4.49(3H,m,H-5,6,6′).ESI-MS(m/z):619.11[M+H]+.
1.2.2 2,3,4,6-四-O-苯甲酰基半乳糖三氯乙酰亚胺酯
1.2.2.1 全苯甲酰基半乳糖的合成
称取1.0 g(5.6 mmol)葡萄糖溶于20 mL无水吡啶,冰浴下缓慢滴加苯甲酰氯5 mL(43.6 mmol),滴加时溶液变浑浊,10 min内滴加完毕.自然升至室温搅拌反应8 h,向反应液中加甲醇2 mL淬灭反应,减压蒸除溶剂,以二氯甲烷溶解剩余物,分别以稀盐酸30 mL,碳酸氢钠30 mL,饱和食盐水30 mL洗涤3次,分离有机相,无水MgSO4干燥,无需纯化直接进行下一步.收率97 %.mp:135~138 ℃.1H-NMR(600 MHz,CDCl3),δ:8.23~7.30(25H,m,Ar—H);6.79(1H,d,J=2.8 Hz,H-1);6.28(1H,t,J=3.6 Hz,H-4);5.75(1H,dd,J=9.6 Hz,3.6 Hz,H-3);4.69(1H,dd,J=2.8 Hz,9.6 Hz,H-2);4.68,4.52,4.46(3H,m,H-5,6,6′).ESI-MS(m/z):700.21[M+H]+.
1.2.2.2 端基的溴代反应
取3.5 g(5.0 mmol)1.2.2.1反应产物,溶于20 mL无水二氯甲烷中,加入质量分数33 % HBr-CH3COOH溶液1.5 mL,溶液变成浅黄色,室温搅拌8 h后在冰浴下加入饱和碳酸氢钠溶液20 mL,分离有机相,有机相用饱和食盐水洗涤,硫酸镁干燥,无需纯化直接进行下一步反应.收率86 %.mp:111~114 ℃.1H-NMR(600 MHz,CDCl3),δ:8.14~7.35(20H,m,Ar—H);6.85(1H,d,J=2.9 Hz,H-1);6.15(1H,dd,J=9.6 Hz,2.9 Hz,H-2);6.28(1H,t,J=3.2 Hz,H-4);5.35(1H,dd,J=9.6 Hz,3.2 Hz,H-3);4.75,5.0,5.15(3H,m,6′,6,5).ESI-MS(m/z):659.50[M+H]+.
1.2.2.3 异头碳水解
取2.5 g(4.1 mmol)干燥的溴代苯甲酰基半乳糖溶于20 mL丙酮中,再加入0.5 mL蒸馏水,加入0.8 g Ag2CO3室温搅拌4 h.加硅藻土抽滤,减压蒸出溶剂丙酮,滴加几滴甲苯将水蒸出,红外灯下干燥.收率82.5 %.mp:85~87 ℃.1H-NMR(600 MHz,CDCl3),δ:8.25~7.30(20H,m,Ar—H);6.05(1H,d,J=3.0 Hz,H-1);5.64(1H,t,J=3.8 Hz,H-4);5.20(1H,dd,J=10.2 Hz,3.0 Hz,H-2);5.33(1H,t,J=10.2 Hz,3.8 Hz,H-3);4.71,4.58,4.42(3H,m,5,6,6′).ESI-MS(m/z):596.23[M+H]+.
1.2.2.4 2,3,4,6-四-O-苯甲酰基半乳糖三氯乙酰亚胺酯
取2.0 g(3.3 mmol)1.2.2.3产物1-羟基苯甲酰半乳糖溶于30 mL无水二氯甲烷,加入DBU 0.48 L,搅拌10 min,加入0.6 mL三氯乙腈,搅拌5 h,蒸干溶剂,剩余物溶解于适量二氯甲烷中,以碱化(三乙胺)的中性氧化铝拌样,自然挥干溶剂.用20倍量碱化的中性氧化铝装柱,以石油醚-乙酸乙酯[V(石油醚)/V(乙酸乙酯)=3/1]淋洗,减压回收溶剂减压蒸干溶剂得目标产物.产率74 %.mp:68~73 ℃.1H-NMR(600 MHz,CDCl3),δ:10.03(1H,s,NH);8.08~7.30(20H,m,Ar—H);6.79(1H,d,J=2.8 Hz,H-1);6.28(1H,t,J=3.6 Hz,H-4);5.82(1H,dd,J=9.6 Hz,3.6 Hz,H-3);4.69(1H,dd,J=2.8 Hz,9.6 Hz,H-2);4.65,4.64,4.49(3H,m,H-5,6,6′).ESI-MS(m/z):619.0[M+H]+.
1.2.3 1,2,3,4,5-五-O-苯甲酰基木糖的合成
1.2.3.1 全苯甲酰木糖的合成
称取0.5 g(2.8 mmol)木糖溶于10 mL无水吡啶,冰浴下缓慢滴加苯甲酰氯3 mL(22.8 mmol).滴加完毕,升至室温搅拌反应5 h,向反应液中加甲醇1 mL淬灭反应,减压蒸除溶剂,以二氯甲烷溶解剩余物,分别以稀盐酸15 mL,碳酸氢钠15 mL,饱和食盐水15 mL洗涤3次,分离有机相,无水MgSO4干燥,无需纯化直接进行下一步.收率95 %.1H-NMR(600 MHz,CDCl3),δ:8.31~7.45(20H,m,Ar—H);6.50(1H,d,J=2.8 Hz,H-1);5.98(1H,t,J=3.2 Hz,H-2);5.85(1H,m,H-3);5.59(1H,m,H-4);4.50(1H,d,J=13.2 Hz,H-5);4.23(1H,dd,J=2.0 Hz,13.2 Hz,H-5′).ESI-MS(m/z):566.1[M+H]+.
1.2.3.2 异头碳的溴代
取1.5 g(2.6 mmol)1.2.3.1反应产物,溶于20 mL无水二氯甲烷中,加入质量分数33 %HBr-CH3COOH溶液1.0 mL,溶液变成浅黄色,室温搅拌5 h后冰浴下加入饱和碳酸氢钠溶液20 mL,分离有机相,有机相用饱和食盐水洗涤,硫酸镁干燥,无需纯化直接进行下一步反应.收率87 %.mp:121~123 ℃.1H-NMR(600 MHz,CDCl3),δ:8.14~7.45(15H,m,Ar—H);6.42(1H,d,J=2.8 Hz,H-1);5.86(1H,t,J=3.2 Hz,H-2);5.62(1H,m,H-3);5.39(1H,m,H-4);4.31(1H,d,J=13.2 Hz,H-5);4.05(1H,dd,J=2.0 Hz,13.2 Hz,H-5′).ESI-MS(m/z):525.3[M+H]+.
1.2.3.3 异头碳水解
取1.2 g(2.2 mmol)干燥的溴代苯甲酰基木糖溶于20 mL丙酮中,再加入0.5 mL蒸馏水,加入0.35 g Ag2CO3室温搅拌4 h.硅藻土抽滤,减压蒸出溶剂,滴加几滴甲苯将水蒸出,红外灯下干燥.收率83 %.mp:135~137 ℃.1H-NMR(600 MHz,CDCl3),δ:8.22~7.45(15H,m,Ar—H);6.22(1H,d,J=3.0 Hz,H-1);5.66(1H,t,J=3.2 Hz,H-2);5.58(1H,m,H-3);5.39(1H,m,H-4);4.11(1H,d,J=11.5 Hz,H-5);3.95(1H,d,J=2.0 Hz,11.5 Hz,H-5′).ESI-MS(m/z):462.6[M+H]+.
1.2.3.4 2,3,4-三-O-苯甲酰基木糖三氯乙酰亚胺酯的合成
取0.8 g(1.7 mmol)1.2.3.3产物1-羟基苯甲酰葡萄糖溶于30 mL无水二氯甲烷,加入DBU 0.48 mL,搅拌10 min,加入0.6 mL三氯乙腈,搅拌5 h,蒸干溶剂,剩余物溶解于适量二氯甲烷中,以碱化(三乙胺)的中性氧化铝拌样,挥干溶剂.用20倍量碱化的中性氧化铝装柱.以石油醚-乙酸乙酯[V(石油醚)/V(乙酸乙酯)=3/1]淋洗,减压回收溶剂蒸干溶剂得目标产物.产率69 %.mp:65~67 ℃.1H-NMR(600 MHz,CDCl3),δ:10.03(1H,s,NH);8.14~7.35(15H,m,Ar—H);6.42(1H,d,J=2.8 Hz,H-1);5.86(1H,t,J=3.2 Hz,H-2);5.62(1H,m,H-3);5.39(1H,m,H-4);4.31(1H,d,J=13.5 Hz,H-5);4.05(1H,d,J=2.0 Hz,13.5 Hz,H-5′).ESI-MS(m/z):605.5[M+H]+.
1.2.4 1,2,3,4-四-O-苯甲酰基吡喃鼠李糖的合成
1.2.4.1 全苯甲酰鼠李糖的合成
称取0.5 g(3.0 mmol)鼠李糖溶于20 mL无水吡啶和无水二氯甲烷混合溶剂中[V(无水吡啶)/V(无水二氯甲烷)=1],冰浴下缓慢滴加苯甲酰氯3 mL(43.6 mmol).滴加完毕,升至室温搅拌反应5 h,向反应液中加甲醇2 mL淬灭反应,减压蒸除溶剂,以二氯甲烷溶解剩余物,分别以稀盐酸20 mL,碳酸氢钠20 mL,饱和食盐水20 mL洗涤3次,分离有机相,无水MgSO4干燥,无需纯化直接进行下一步.收率95 %.mp:153~155 ℃.1H-NMR(600 MHz,CDCl3),δ:8.25~7.30(20H,m,Ar—H);6.58(1H,d,J=2.8 Hz,H-1);5.85(1H,t,J=3.1 Hz,H-4);5.65(1H,dd,J=2.8 Hz,9.5 Hz,H-2);5.58(1H,t,J=9.5 Hz,3.1 Hz,H-3);4.39(1H,q,J=6.2 Hz,H-5);1.16(3H,d,J=6.2 Hz,CH3).ESI-MS(m/z):580.5[M+H]+.
1.2.4.2 异头碳溴代
取1.2 g(2.5 mmol)1.2.4.1反应产物,溶于20 mL无水二氯甲烷中,加入质量分数33 %HBr-CH3COOH溶液1.5 mL,溶液变成浅黄色,室温搅拌8 h后在冰浴下加入饱和碳酸氢钠溶液20 mL,分离有机相,有机相用饱和食盐水洗涤,硫酸镁干燥,无需纯化直接进行下一步反应.收率87 %.mp:132~134 ℃.1H-NMR(600 MHz,CDCl3),δ:8.05~7.30(15H,m,Ar—H);6.44(1H,d,J=3.0 Hz,H-1);5.81(1H,t,J=2.5 Hz,H-4);5.73(1H,dd,J=3.0 Hz,10.4 Hz,H-2);5.62(1H,dd,J=10.4 Hz,2.5 Hz,H-3);4.43(1H,q,J=5.6 Hz,H-5);1.22(3H,d,J=5.6 Hz,CH3).ESI-MS(m/z):539.5[M+H]+.
1.2.4.3 异头碳水解
取1.1 g(2.0 mmol)干燥的溴代苯甲酰基鼠李糖溶于20 mL丙酮中,再加入0.5 mL蒸馏水,加入0.3 g Ag2CO3室温搅拌4 h.加硅藻土抽滤,减压蒸出溶剂丙酮,滴加几滴甲苯将水蒸出,红外灯下干燥.收率86 %.mp:99~103 ℃,1H-NMR(600 MHz,CDCl3),δ:8.10~7.34(15H,m,Ar—H);6.44(1H,s,H-1);5.81(1H,s,H-4);5.73(1H,dd,J=3.0 Hz,10.0 Hz,H-2);5.62(1H,t,J=8.9 Hz,2.5 Hz,H-3);4.39(1H,q,J=6.2 Hz,H-5);1.22(3H,d,J=6.2 Hz,CH3).ESI-MS(m/z):476.5[M+H]+.
1.2.4.4 2,3,4-三-O-苯甲酰基鼠李糖三氯乙酰亚胺酯的合成
取0.8 g(1.6 mmol)1.2.4.3上一步产物1-羟基苯甲酰鼠李糖溶于30 mL无水二氯甲烷,加入DBU 0.48 mL,搅拌10 min,加入0.6 mL三氯乙腈,搅拌5 h,蒸干溶剂,剩余物溶解于适量二氯甲烷中,以碱化(三乙胺)的中性氧化铝拌样,自然挥干溶剂.用20倍量碱化的中性氧化铝装柱.以石油醚-乙酸乙酯[V(石油醚)/V(乙酸乙酯)=3/1]淋洗,减压回收溶剂减压蒸干溶剂得目标产物.产率79 %.mp:83~91 ℃.1H-NMR(600 MHz,CDCl3),mp:85~86 ℃,δ:10.15(1H,s,NH);8.00~7.34(15H,m,Ar—H);6.44(1H,s,H-1);5.81(1H,s,H-4);5.73(1H,dd,J=3.0 Hz,10.0 Hz,H-2);5.62(1H,dd,J=10.0 Hz,2.6 Hz,H-3);4.39(1H,q,J=6.2 Hz,H-5);1.32(3H,d,J=6.2 Hz,CH3).ESI-MS(m/z):619.4[M+H]+.
1.3 齐墩果酸苄酯的合成
称量0.20 g(0.44 mmol)齐墩果酸,溶于DMF中,加入溴苄0.5 mL,无水碳酸钾0.06 g,室温反应.TLC检测反应终点,展开剂[V(石油醚)/V(乙酸乙酯)=3/1],10 h反应结束.以饱和食盐水打浆,过滤后得白色产物0.18 g,产率为75 %.mp:254.5~256.3 ℃.1H-NMR(600 MHz,CDCl3),δ:7.3~7.89(m,5H,Ar—H);5.21(s,1H,H-12);2.74(t,J=7.5 Hz,1H,H-18);5.34(s,2H,PhCH2);2.96(m,1H,H-3);2.01~1.21(m,25H);1.02(s,3H,CH3);0.97(s,3H,CH3);1.20(s,3H,CH3);1.10(s,3H,CH3);0.99(s,3H,CH3);0.89(s,3H,CH3);0.87(s,3H,CH3).ESI-MS(m/z):546.5[M+H]+.
1.4 齐墩果酸葡萄糖皂苷Ⅰa的合成
1.4.1 糖苷化反应
取0.1 g(1.8 mmol)糖苷受体,0.15 g苷元,溶于无水二氯甲烷中,加0.3 g 4A分子筛,冰盐浴下搅拌0.5 h,加5滴TMSOTf,冰浴下反应4 h,加几滴三乙胺淬灭反应,过滤,减压蒸除溶剂,无需纯化直接进行下一步反应.收率75 %.mp:201.5~205.2 ℃.1H-NMR(600 MHz,CDCl3),δ:8.16~7.30(25H,m,Ar—H);5.20(1H,s,H-12);5.54(1H,s,H-1′);5.25(1H,t,J=10.2 Hz,H-3′);4.65(1H,t,J=10.2 Hz,H-4′);4.98(1H,dd,J=3.6 Hz,10.2 Hz,H-2′);4.65,4.64,4.49(3H,m,H-5′,6′,6′);2.79~3.01(m,1H,H-3);1.98~1.21(m,25H,1.10(s,3H,CH3);0.97(s,3H,CH3);1.05(s,3H,CH3);1.16(s,3H,CH3);0.99(s,3H,CH3);0.89(s,3H,CH3);0.87(s,3H,CH3).ESI-MS(m/z):1125.5[M+H]+.
1.4.2 水解反应
取1.4.1的反应产物3-O-(β-D-2,3,4,6-四-O-苯甲酰基吡喃葡萄糖)齐墩果烷型-11烯-28-苄酯,溶于已配好的甲醇钠/甲醇溶液[13](pH≈10)中.TLC检测反应终点,展开剂V(石油醚)/V(乙酸乙酯)=6/1,4 h反应结束.反应液中再加入适量阳离子交换树脂搅拌,调节pH值使溶液呈中性.过滤,减压蒸除溶剂.粗品经硅胶柱色谱分离纯化,洗脱剂V(石油醚)/V(乙酸乙酯)=20/1~10/1,得产物0.62 g.产率67.25 %.mp:221.5~226.2 ℃.1H-NMR(600 MHz,CDCl3),δ:7.28~7.93(m,5H,Ar—H);5.30(s,1H,H-12);5.04(d,J=7.7 Hz,1H,H-1′);2.28(t,J=7.6 Hz,1H,H-18);2.96(m,1H,H-3);3.51~3.47(m,H-2′,3′,5′,3H);3.65(m,1H,H-4′);3.32~3.18(m,H-6′);2.15~1.25(m,25H);1.01(s,3H,CH3);1.10(s,3H,CH3);0.97(s,3H,CH3);0.89(s,3H,CH3);1.03(s,3H,CH3);0.97(s,3H,CH3);1.05(s,3H,CH3).ESI-MS(m/z):708.5[M+H]+.
1.5 3-O-(β-D-吡喃半乳糖)-齐墩果烷型-12-烯-28苄酯Ⅰb的合成
参照Ⅰa合成方法,合成化合物Ⅰb.粗品经硅胶柱色谱纯化,洗脱剂V(石油醚)/V(乙酸乙酯)=20/1~10/1,得白色固体,收率为65.12 %.mp:213.1~217.0 ℃.1H-NMR(600 MHz,CDCl3),δ:7.85~7.46(m,5H,Ar—H);5.26(s,1H,H-12);4.95(d,J=7.5 Hz,1H,H-1′);2.18(t,J=6.7 Hz,1H,H-18);2.88(m,1H,H-3);3.51(m,1H,H-4′);3.40~3.23(m,H-2′,3′,5′,3H);2.01~1.36(m,25H);1.09(s,6H,2CH3);1.16(s,6H,2CH3);0.98(s,3H,CH3);0.90(s,3H,CH3);1.15(s,3H,CH3).ESI-MS(m/z):708.5[M+H]+.
1.6 3-O-(α-D-吡喃木糖)-齐墩果烷型-12-烯-28-苄酯Ⅰc的合成
参照Ⅰa合成方法,合成化合物Ⅰc.粗品经硅胶柱色谱纯化,洗脱剂V(石油醚)/V(乙酸乙酯)=20/1~10/1,得白色固体,收率58.25 %.mp:194.7~197.2 ℃.1H-NMR(600 MHz,CDCl3),δ:7.84~7.43(m,5H,Ar—H);5.27(s,1H,H-12);4.89(d,J=2.8 Hz,1H,H-1′);2.26(t,J=7.5 Hz,1H,H-18);2.78(m,1H,H-3);3.55~3.33(m,H-2′,3′,4′,3H);3.26~3.11(m,2H,H-5,5′);1.08(s,6H,2CH3);1.02(s,3H,CH3);0.98(s,3H,CH3);0.87(s,3H,CH3);1.15(s,6H,2CH3);1.98~1.29(m,25H).ESI-MS(m/z):678.4[M+H]+.
1.7 3-O-(α-L-吡喃鼠李糖)-齐墩果烷型-12-烯-28苄酯Ⅰd的合成
参照Ⅰa的合成方法,合成化合物Ⅰd.粗品经硅胶柱色谱纯化,洗脱剂V(石油醚)/V(乙酸乙酯)=20/1~10/1,得白色固体,收率62.35 %.mp:197.5~203.2 ℃.1H-NMR(600 MHz,CDCl3),δ:7.99~7.33(m,5H,Ar—H);5.27(s,1H,H-12);4.79(d,J=3.2 Hz,1H,H-1′);2.30(t,J=7.6 Hz,1H,H-18);2.90(m,1H,H-3);3.55~3.33(m,H-2′,3′,4′,3H);1.18(d,J=6.1,3H,CH3);1.11(s,6H,2CH3);1.01(s,3H,CH3);0.96(s,3H,CH3);0.89(s,3H,CH3);1.25(s,6H,2CH3);1.95~1.30(m,25H).ESI-MS(m/z):692.4[M+H]+.
2 实验结果与讨论
2.1 化学部分
以齐墩果酸为先导化合物,将C-28位羧基与溴苄反应成酯,A环C-3位羟基进行糖苷化反应,设计并合成了4个齐墩果酸糖苷衍生物.以上目标产物在反应过程中采用TLC监测反应终点,柱层析纯化,其结构通过1H-NMR,MS等得以确证.
糖基三氯乙酰亚胺酯是寡糖及糖缀合物合成中的重要糖苷化试剂[11],然而因为还原性单糖有很多活性羟基,制备糖苷供体过程中主要问题在于异头碳区域选择性反应反应试剂的选择,有文献报道用苄胺[12],乙二胺等弱碱催化异头碳苯甲酰酯的水解,实验发现这些条件会导致其他位置酯基的水解,副产物多,实验条件不易控制.采用氢溴酸-乙酸试剂先溴代后水解,主要是利用异头碳在阳离子引发剂作用下酯键断裂形成能量不是很高的氧鎓离子中间体,从而使反应更有选择性,其次,溴代反应需控制体系无水,因为在阳离子引发剂作用下,若体系有水,根据软硬酸碱理论,水分子会先与羰基碳正离子作用成键,而酯键断裂是速控步,反应慢,因此若体系有水,该歩产率将会很低.
苯甲酰基脱保护[13]步由于产物中有氧苷键,酸催化脱保护容易使苷键断裂,利用氧苷键周围的大位阻环境,在碱性条件下不易进攻苷键使其断裂,故选用甲醇钠/甲醇催化脱保护,并控制pH值在10左右,获得很好的收率.且C-28位苄酯也在此条件下也因为空间位阻的影响不易水解从而得以保存.
2.2 生物活性部分
以吉菲替尼和阿霉素为阳性对照药,采用MTT法对目标化合物进行初步的体外活性测试,活性数据见表1.

表1 目标化合物对Hela、BGC-823细胞的抑制率Table 1 Inhibitory activity of the target compounds on the Hela and BGC-823 cells
3 结果与讨论
从表1所测结果分析:在OA的C-3位联接单糖,C-28位成苄酯得到的衍生物对Hela、BGC-823细胞都具有一定的抗肿瘤活性;Ⅰc、Ⅰd对两种肿瘤细胞的抑制率高于Ⅰa、Ⅰb,可能是因为C-3位连接的脱氧单糖使目标化合物具有更好的活性.由表1可知:本文合成的齐墩果酸糖苷衍生物对Hela和BGC-823细胞均有一定程度的抑制作用,但对BGC-823细胞的抑制作用更加明显.
相对于OA,Ⅰa~Ⅰd对子宫颈癌细胞Hela细胞系和人胃腺癌细胞BGC-823细胞系抑制率更高.在齐墩果酸C-3位连接单糖基增加了齐墩果酸衍生物的亲水性基团,C-28位连接溴苄成酯提高熊果酸衍生物的脂溶性,使其具有良好的脂水分配系数可能是这些衍生物具有较高抑制活性的原因.
这一研究结果为齐墩果酸的进一步结构优化,寻找更高活性的目标分子提供了一定的参考依据.
:
[1] 张明发,沈雅琴.齐墩果酸和熊果酸保肝药理作用的研究进展[J].抗感染药学,2012,9(1):13-19.
[2] 赵龙铉,沈平,郑昌吉,等.齐墩果酸衍生物的合成与表征及其抑菌活性的研究[J].辽宁师范大学学报(自然科学版),2012,35(1):63-68.
[3] 吴悠,张秋,王瑶.齐墩果酸的药理作用机制及其在龋病治疗中的研究进展[J].泸州医学院学报,2014,37(5):544-546.
[4] 赵龙铉,贺兴隆,金礼吉,等.齐墩果酸和甘草次酸衍生物的合成与表征及抗癌活性研究[J].辽宁师范大学学报(自然科学版),2010,33(4):474-479.
[5] 曲正义,刘宏群,郑培和,等.齐墩果酸型皂苷药理活性研究[J].中成药,2012,34(6):1143-1147.
[6] 苏春华,程克光,初相伍,等.齐墩果酸-α-氨基膦酸酯衍生物的合成与抗肿瘤活性研究[J].中国实验方剂学杂志,2014,20(8):123-128.
[7] MALLAVADHANI U V,MAHAPATRA A,PATTNAIK B,et al.Synthesis and Anti-Cancer Activity of Some Novel C-17 Analogs of Ursolic and Oleanolic Acids[J].Medicinal Chemistry Research,2013,22(3):1263-1269.
[8] MATSUDA H,LI Y,MURAKAMI T,et al.Antidiabetic Principles of Natural Medicines.Ⅲ.Structure-Related Inhibitory Activity and Action Mode of Oleanolic Acid Glycosides on Hypoglycemic Activity[J].Chemical & Pharmaceutical Bulletin,1998,46(9):1399-1403.
[9] CATENI F,BONIVENTO P,PROCIDA G,et al.Chemoenzymatic Synthesis and Antimicrobial Activity Evaluation of Monoglactosyl Diglycerides[J].Eur.J.Med.Chem.,2008,43(1):210-221.
[10] 李晓东,康帅涛,许环军,等.糖苷化试剂-糖基三氯乙酰亚胺酯的高效制备[J].沈阳药科大学学报,2011,28(9):707-711.
[11] SCHMIDT R R,KINZY W.Anomeric-Oxygen Activation for Glycoside Synthesis:the Trichloroacetimidate Method[J].Advances in Carbohydrate Chemistry&Biochemistry,1994,50:21-123.
[12] KELLER-JUSLÉN C,KUHN M,STHELIN H,et al.Synthesis and Antimitotic Activity of Glycosidic Lignan Derivatives Related to Podophyllotoxin[J].Journal of Medicinal Chemistry,1971,14(10):936-940.
[13] 庞思平,于永忠.苯甲酰基的脱去[J].合成化学,2001,9(5):407-412.
猜你喜欢
化学工程师(2022年3期)2022-04-19
当代水产(2021年10期)2022-01-12
食品安全导刊(2021年20期)2021-08-30
股市动态分析(2021年8期)2021-04-26
上海化工(2021年2期)2021-04-23
大众健康(2019年9期)2019-10-11
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年5期)2018-06-13
中国药房(2017年13期)2017-05-16
科技与创新(2015年20期)2015-10-29
