
Rh2O3/monoclinic CePO4 composite catalysts for N2O decomposition and CO oxidation☆
2018-05-25 07:50HuanLiuZhenMa
Huan Liu,Zhen Ma*
Shanghai Key Laboratory of Atmospheric Particle Pollution and Prevention,Department of Environmental Science and Engineering,Fudan University,Shanghai 200433,China
1.Introduction
Heterogeneous catalysts are mostly composite materials,i.e.,they in most cases comprise at least two components.One typical case is supported catalysts,in which active components(e.g.,metal,metal oxide)are finely dispersed on solid supportsthat areoften metal oxides,SiO2,and zeolites.However,metal salts are traditionally not often used as catalyst supports,but LaPO4and hydroxyapatite(Ca10(PO4)6(OH)2)have been used to make supported catalysts such as Au/LaPO4[1–5],Pt/LaPO4[6,7],Pd/LaPO4[8],Rh/LaPO4[9–11],Ru/LaPO4[12],Au/hydroxyapatite[13–18],Rh/hydroxyapatite[10,19,20],and Ru/hydroxyapatite[21–25].The development of metal phosphate-supported catalysts is interesting,not only because such catalysts are limited in number,but also because these catalysts combine the acid–base and/or redox functions of the supports and the catalytic functions of the supported metals or metal oxides.
CePO4is similar to LaPO4,i.e.,they both belong to lanthanide phosphates.CePO4can be used as a catalyst for the oxidative dehydrogenation of is obutane[26,27],amination of 1-octanol[28],vapor-phase O-alkylation of phenol[29],dehydration reaction of 2-propanol[30],and selective catalytic reduction of NO with NH3[31,32].It can also be used to make supported catalysts such as Ru/CePO4for the aerobic oxidation of alcohols[33],Pt/CePO4for the selective catalytic reduction activity of NOx[34],and Au/CePO4for CO oxidation[35].However,examples on CePO4-supported catalysts and their catalytic applications are still limited in number.
CePO4presents two phases:hexagonal and monoclinic.Hexagonal CePO4nanoparticles can be prepared by direct precipitation[36,37],and hexagonal CePO4nanowirescan beprepared via hydrothermal process[38,39].Monoclinic CePO4can be prepared by calcining hexagonal CePO4at high temperatures[40,41].Ru/CePO4[33],Pt/CePO4[34],and Au/CePO4[35]mentioned above all contain hexagonal CePO4supports,but monoclinic CePO4-based catalysts have been rarely reported.
N2O is an environmental pollutant with a green-house effect and may contribute to the ozone layer depletion.Catalysts used for the decomposition of N2O include metal oxides,ion-exchanged zeolites,and supported metal catalysts[42–44].In particular,some Rh-based catalysts showsuperior performance.Rh-based catalysts for N2O decomposition include Rh-ZSM-5[45],Rh/MgO[46],Rh/USY[47],Rh/Al2O3[48,49],Rh/CeO2[50,51],Rh/KIT-6[52],Rh/SBA-15[53,54],Rh/MCM-41[55],Rh2O3/LaPO4[10,11],and Rh2O3/hydroxyapatite[10,19,20].It would be interesting to explore other metal phosphate-based Rh2O3catalysts.
In this work,we report Rh2O3supported on monoclinic CePO4nanoparticles and nanowires as well as the catalytic performance in N2O decomposition and CO oxidation.It was found that Rh2O3supported on monoclinic CePO4nanowires is much more active than Rh2O3supported on monoclinic CePO4nanoparticles.Reasons for these observations were elucidated through detailed characterization.
2.Experimental
2.1.Preparation
2.1.1.Monoclinic CePO4nanoparticles[36,40]
6.95 g Ce(NO3)3·6H2O(Aladdin,99.9%)and 1.84 g NH4H2PO4(Sinopharm,AR)were dissolved in 80 ml deionized water under magnetic stirring for 20 min.The p H value was adjusted to 7 by adding aqueous ammonia(27 wt%).The mixture was stirred continuously for 6 h.The precipitate was filtered,washed with ethanol,and dried at 80°C.The product(hexagonal CePO4nanorods)was calcined at 900°C for 3 h to form monoclinic CePO4nanoparticles(CePO4-MNP).
2.1.2.Monoclinic CePO4nanowires[40]
6.95 g Ce(NO3)3·6H2Oand 1.84 g NH4H2PO4were dissolved in 80 ml deionized water under magnetic stirring for 20 min.The p H value was adjusted to 1 by adding aqueous ammonia(27 wt%).The suspension was transferred into a Te flon-lined stainless steel autoclave and purged with flowing He for 1 h to prevent the oxidation of Ce3+to Ce4+before heating.After that,the suspension was heated at 150°C for 12 h in an oven.The precipitate was filtered,washed with ethanol,and dried at 80°C.The product(hexagonal CePO4nanowires)was calcined at 900°C for 3 h to form monoclinic CePO4nanowires(CePO4-MNW).
2.1.3.Rh2O3/CePO4catalysts
2 ml Rh(NO3)3solution(0.01 g·ml−1based on Rh)was placed in a agate mortar containing 1.98 g LaPO4.The mixture was ground till dry under an infrared lamp.The final powders were calcined at 500°Cfor 3 h.
2.2.Characterization
XRD data were collected on a MSALXD2 instrument under the conditions of:2θ =10°–80°,scanning rate 4(°)·min−1,and scanning step 0.01°.The specific BET surface areas were measured by N2adsorption–desorption at−196 °C on a Micromeritics Tris tar 3000 instrument.ICP-OES data on the Rh contents were obtained using a Perkin-Elmer OPTIMA 2100 DV optical emission spectrometer,following the procedure described elsewhere[56].
TEM images were obtained on a JEM-2011F instrument coupled with EDS[11].The sizes of Rh2O3nanoparticles were obtained by measuring 100 particles for each sample,using the Digital Micrograph software.
X-ray photoelectron spectroscopic(XPS)data were recorded on a Perkin-Elmer PHI 5000 C spectrometer with Mg KαX-ray source.The binding energies(BE)were corrected by C1s peak(284.8 eV).CASXPS software was used for analysis.
CO2temperature-programmed desorption(CO2-TPD)experiments were performed on a FINESORB-3010 instrument[10,12].0.15 g of sample was pretreated at 200 °C with He(30 ml·min−1),and then cooled down to 50°C.The sample was saturated with 5%CO2/He(40 ml·min−1)for 1 h and swept by He flowat 50 °Cfor 3 h.The sample was then heated to 600°Cat aramping rate of 10°C·min−1.The amount of desorbed CO2was calibrated using 5%CO2/He with known volume.
O2temperature-programmed desorption(O2-TPD)experiments were carried out on a FINESORB-3010 instrument[10,12].A sample(0.15 g)was pretreated with He(30 ml·min−1)at 500 °C for 1 h,and cooled to 50 °C.Then O2(30 ml·min−1) flowed through the sample at 50°C for 1 h.After that,the system was purged by flowing He(30 ml·min−1)for 3 h,and the sample was then heated from 50 to 600 °C with a heating rate of 10 °C·min−1.
Temperature-programmed reduction (H2-TPR) experiments were carried out on a self-built instrument.50 mg of sample was pretreated at 200 °C with N2(30 ml·min−1)for 1 h and then cooled down to 50 °C.10%H2/N2(30 ml·min−1) flowed through the sample for 2–3 h until the baseline reached stable,and then the sample was heated to 350 °C at a ramping rate of 5 °C·min−1.H2consumption was analyzed by FULI 9750 gas chromatograph.
2.3.Catalytic activity measurements
N2O decomposition was studied in a fixed bed reactor.0.25 g of catalyst was loaded into a U-shaped quartz tube(7 mm diameter)[20,57].0.5%N2O/He( flow rate:60 ml·min−1)was introduced into the quartz tube.The catalyst was maintained at room temperature for 1 h,and the reaction temperature was then increased stepwise and kept at each temperature step for 0.5 h.The effluent gas was analyzed every 10 min using an on-line GC(Agilent 7890A).The N2O conversion was calculated as([N2O]in−[N2O]out)/[N2O]in×100%.
The effect of co-fed CO2,O2,H2O and O2/H2O vapor on the catalytic activity was studied by adding 2%CO2,5%O2,or 2%H2O into there action mixture whereas the concentration of N2O was still 0.5%,and the total flow rate of the gas was still 60 ml·min−1.
Catalytic CO oxidation was studied in a fixed bed reactor described elsewhere[5].The reaction conditions are:0.25 g catalyst in a U-shaped quartz tube(5 mm inner diameter),1%CO in air, flow rate 50 ml·min−1.The catalyst was maintained at room temperature for 1 h in the presence of flowing 1%CO,and heated to 200°C with a ramping rate of 0.5 °C·min−1.The effluent gas was analyzed every 10 min using an on-line gas chromatograph(GC;Agilent 7890A,equipped with a TCD detector).The CO conversion was calculated as([CO]in−[CO]out)/[CO]in×100%.
3.Results
Fig.1 shows the XRD patterns of CePO4and Rh2O3/CePO4.CePO4-MNP[36,40]and CePO4-NMW[38,40]prepared according to the methods in the literature both show monoclinic phase(PDF#32-0199).The XRD patterns of Rh2O3/CePO4samples are similar to those of their corresponding supports.No Rh2O3phase can be detected,probably due to the lowRh content(~1 wt%).
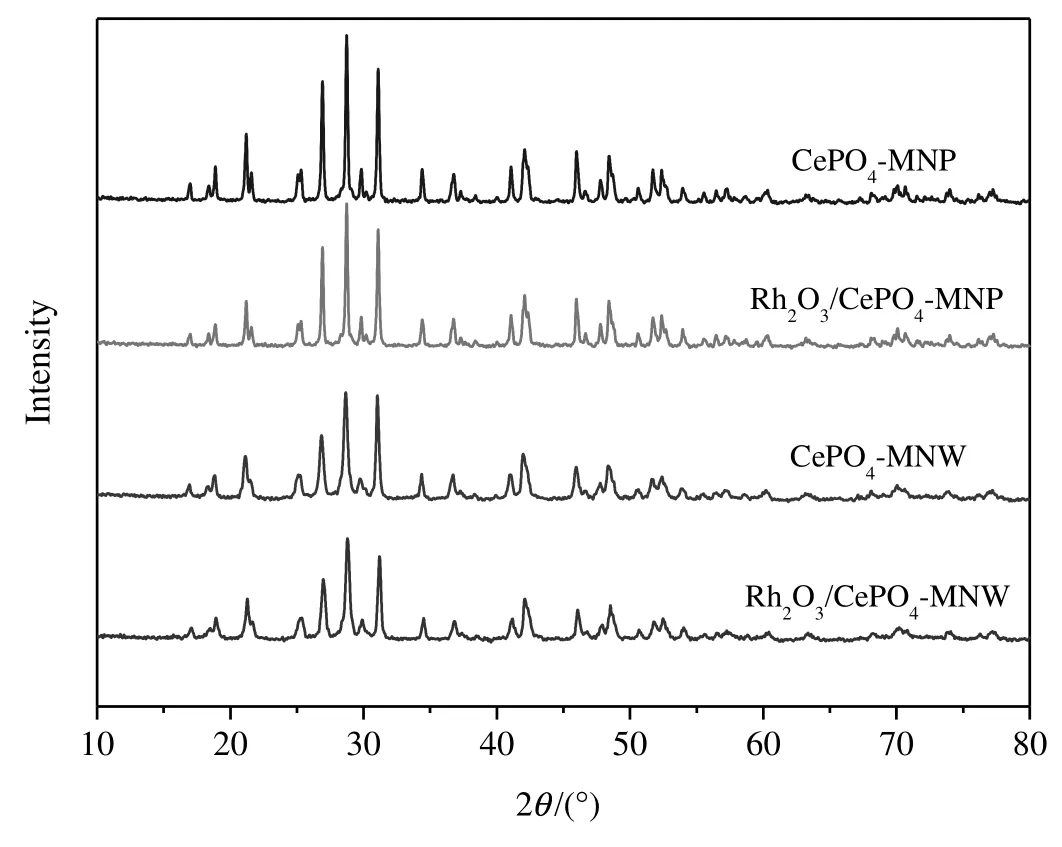
Fig.1.XRD patterns of Rh2O3/CePO4 catalysts.
Fig.2 shows the TEM and HRTEM images of Rh2O3/CePO4catalysts.Rh2O3/CePO4-MNP is primarily composed of large CePO4nanoparticles with sizes of 40–200 nm.Rh2O3/CePO4-MNW exhibits CePO4nanowires with widths of 15–80 nm and lengths of 100–900 nm.The interplanar distances of CePO4-MNP and CePO4-MNW are 0.602 and 0.606 nm,respectively,corresponding to the(100)plane of monoclinic CePO4[58].

Fig.2.TEM and HRTEM images of Rh2O3/CePO4 catalysts.
Supported Rh2O3particles on CePO4supports can be seen clearly in Figs.S1–S2.On the basis of 100 particles for each sample,the mean Rh2O3particle sizes are estimated to be(3.0±0.9)nm and(2.5±0.7)nm for Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW,respectively.The difference in Rh2O3particle sizes may be due to the nature of supports,i.e.,the BET surface areas of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are <1 m2·g−1and 16.6 m2·g−1,respectively.The fact that the surface area of Rh2O3/CePO4-MNP is lower than that of Rh2O3/CePO4-MNW is consistent with the fact that the XRD peaks of Rh2O3/CePO4-MNP are much sharper than the corresponding XRD peaks of Rh2O3/CePO4-MNW(Fig.1).In general,support with a larger surface area tends to better disperse the supported active component and make its size smaller.
3.1.Catalytic performance
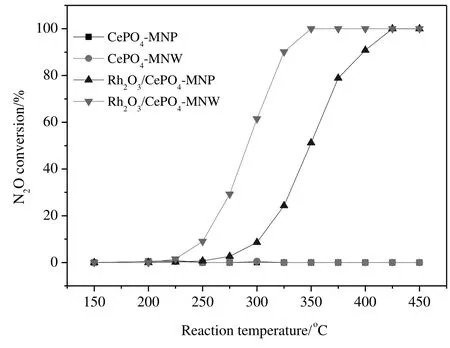
Fig.3.N2O conversions over Rh2O3/CePO4 catalysts as a function of reaction temperature.
Fig.3 shows N2O conversion over Rh2O3/CePO4catalysts.The T50(temperature required for reaching 50%conversion)values of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are 348 and 291°C,respectively,indicating that the latter is much more active than the former.The N2O conversions of these catalysts at 325°C are 24.3%and 90.1%,respectively.The actual Rh contents on CePO4-MNP and CePO4-MNW are determined by ICP-OES as 1.02 wt%and 0.99 wt%,respectively.Thus,the specific rates of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW at 325 °C are calculated to be 77 and 292 mmol·(g Rh)−1·h−1,respectively,meaning that Rh2O3/CePO4-MNW is significantly more active than Rh2O3/CePO4-MNP.The corresponding CePO4supports showno activity below400°C.
The stability of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW as a function of reaction time on stream was tested at 400 and 325°C,respectively.As shown in Fig.4,the initial N2O conversions(90%and 96%for Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW,respectively)are consistent with the N2O conversion read out from the conversion curves in Fig.3.After 40 h on stream,the N2O conversions on Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are 84%and 92%,respectively,i.e.,both catalysts are relatively stable.
We additionally tested the Influence of co-fed CO2,O2,H2O,or O2/H2O on catalytic activity.As shown in Fig.5,2%CO2has no effect on the activity of the two catalysts.The catalytic activities of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are only slightly inhibited in the presence of 5%O2,but the presence of 2%H2O seriously inhibits the catalytic activity,as the T50values of these catalysts increase by 87 °C and 88 °C,respectively.The obvious inhibiting effect is due to the competitive adsorption of H2O[10,12].The catalytic activity is further inhibited in the presence of both O2and H2O,as the T50values of these catalysts increase by 89 °C and 113 °C,respectively.
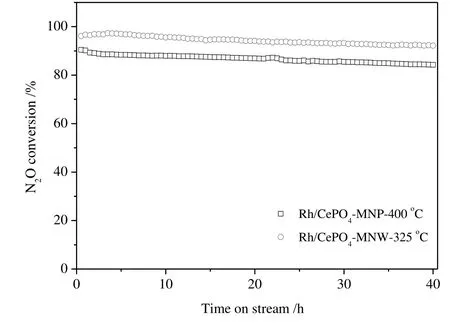
Fig.4.N2O conversions over Rh2O3/CePO4-MNP(reaction temperature:400°C)and Rh2O3/CePO4-MNW(reaction temperature:325°C),as a function of time on stream.
The inhibiting effect and its reversibility were studied by isothermal gas-switching experiments.As shown in Fig.6a,the N2O conversion on Rh2O3/CePO4-MNP is about 91%at 400°C in the first step,consistent with the value seen in Fig.3.The N2O conversion decreases to 79%when 5%O2is introduced.By removing O2,the N2O conversion returns to 86%.After two cycles,the N2O conversion decreases by 8%.The same trend is found with the introduction and retraction of 2%H2O.The data indicate that the inhibiting effect of O2and H2Oon Rh2O3/CePO4-MNPis reversible.

Fig.5.The Influence of co-feed 2%CO2,5%O2,2%H2Oor 5%O2+2%H2Oon the conversion of N2O over Rh2O3/CePO4-MNP(a)and Rh/CePO4-MNW(b).

Fig.6.N2O conversions over Rh2O3/CePO4-MNP in the absence or presence of 5%O2 or 2%H2O at 400°C.
Fig.7 shows the effect of co-fed 5%O2or 2%H2O on the performance of Rh2O3/CePO4-MNW at 325°C.The introduction of 5%O2or 2%H2O can cause a sudden drop of N2O conversion,but the catalytic activity can be completely restored after removing 5%O2or 2%H2O in two cycles.
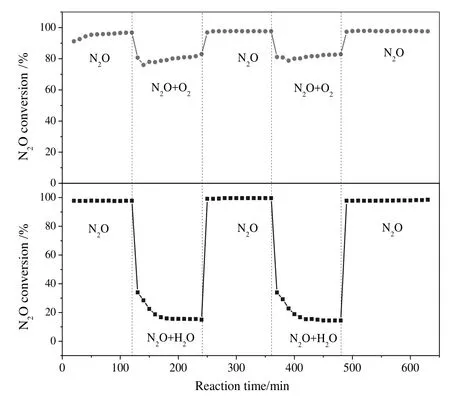
Fig.7.N2O conversion over Rh2O3/CePO4-MNW in the absence or presence of 5%O2 or 2%H2O at 325°C.
The stability of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW as a function of reaction time on stream was also tested at 450°C in the presence of 5%O2and 2%H2O.As shown in Fig.S3,the N2O conversion at 450°C increases slightly from 62%to 65%on Rh2O3/CePO4-MNP during the initial 2 h.After 40 h of continuous reaction,the N2O conversion decreases slightly to 59%.On Rh2O3/CePO4-MNW,the N2O conversion is maintained at 97%after 40 h of continuous reaction.TEM characterization of the spent Rh2O3/CePO4-MNP(Fig.S4)and Rh2O3/CePO4-MNW(Fig.S5)shows that the mean Rh2O3particle sizes on these catalysts are(3.0±0.7)nm and(2.5±0.6)nm,respectively.
These catalysts were also tested in CO oxidation because it is a conventional and sensitive probe reaction.As shown in Fig.S6,Rh2O3/CePO4-MNW is still more active than Rh2O3/CePO4-MNP.The CO conversionsover these two catalystsat 90°Care9.1%and 38.3%,respectively.For comparison,the CO conversions achieved over Rh2O3/hexagonal CePO4nanorods and Rh2O3/hexagonal CePO4nanowires at 90°C are 65.1%and 86.6%,respectively(Fig.S7).The data show that the advantage of CePO4nanowires over CePO4nanorods/nanoparticles in making supported Rh2O3catalysts for CO oxidation is general,i.e.,regardless of hexagonal or monoclinic phase.
The stability of Rh2O3/CePO4-MNW and Rh2O3/CePO4-MNP was tested at 100 and 145°C,respectively(Fig.S8).The catalytic activity of two catalysts decreases rapidly in the initial 2 h,probably due to the accumulation of carbonate/formate on catalyst surfaces.After that,the catalytic activity continues to decline to 28%on Rh2O3/CePO4-MNP,but it can be maintained at around 65%on Rh2O3/CePO4-MNW.
首先,思政课教师的思想激励作用。“工匠精神培育的最佳时机就在高职生入学的开始一二年,这段时间也正是公共必修思政课的开课时期,把工匠精神的培育有效地融入高职思政教育必然有助于增加具有工匠精神人才的有效供给,为我国制造业的升级转型培养更多具有高级职业精神和高尚爱国主义情操的高素质人才。[5]”思政课要有针对性,能够解决学生在学习生活中的困难和疑虑,至少能给学生一种直面问题的思路和方法。学生在专业的学习上会遇到各种各样的困难,思政课教师要从思想上发挥答疑解惑的作用,引导学生去面对困难,用勇气解决困难。这是培养学生工匠精神必须具备的思路。
3.2.Additional characterization of CePO4 and Rh2O3/CePO4 catalysts
Fig.S9 shows the Rh 3d XPS spectra Rh2O3/CePO4catalysts.The binding energies of two catalysts locate at 306–311 and 311–318 eV,assigned to Rh3+3d5/2and Rh3+3d3/2[59,60],respectively.The data confirm that the Rh species exist as Rh2O3.
Fig.S10 shows the Ce 3d XPS spectra of Rh2O3/CePO4catalysts.The spectra display two multiplets assigned to the spin orbit split 3d5/2and 3d3/2core holes[31],respectively.The doublet peaks at 880.5–881.5 and 898.8–899.5 eV correspond to Ce(III)3d94f1O2p6.The doublet peaks at 884.4–884.9 and 902.8–903.5 eV are assigned to Ce(III)3d94f2O2p5[61,62].The data prove that cerium exists as Ce3+,with no Ce4+in both samples.
Fig.S11 shows the O 1s spectra of Rh2O3/CePO4catalysts.The main peaks at ca.530.4 eV correspond to lattice oxygen and a shoulder peak at ca.532.5 eV is attributed to hydroxyl groups[31,63].The ratio of hydroxyl groups(among all oxygen species)in Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW is similar(11.6%and 11.8%,respectively).

Fig.8.CO2-TPD pro files of Rh2O3/CePO4 catalysts.
Fig.8 shows the CO2-TPD pro files of CePO4and Rh2O3/CePO4.CO2desorption peak is not detected on CePO4-MNP and Rh2O3/CePO4-MNP.For comparison,CePO4-MNW and Rh2O3/CePO4-MNW both exhibit a CO2desorption peak ranging from ~150 °C to ~450 °C.The amounts of basic sites of CePO4-MNW and Rh2O3/CePO4-MNW are 31 and 28 μmol·g−1,respectively.The data indicate that the basicity of catalysts mainly originates from the supports.
Fig.9 shows the O2-TPD pro files of CePO4and Rh2O3/CePO4.CePO4-MNP and Rh2O3/CePO4-MNP do not showany O2desorption peak,indicating that O2does not adsorb on CePO4-MNP and Rh2O3/CePO4-MNP at 50°C.CePO4-NMW and Rh2O3/CePO4-MNW both exhibit a broad O2desorption peak,and the desorption of O2starts at 129 and 133°C,respectively.
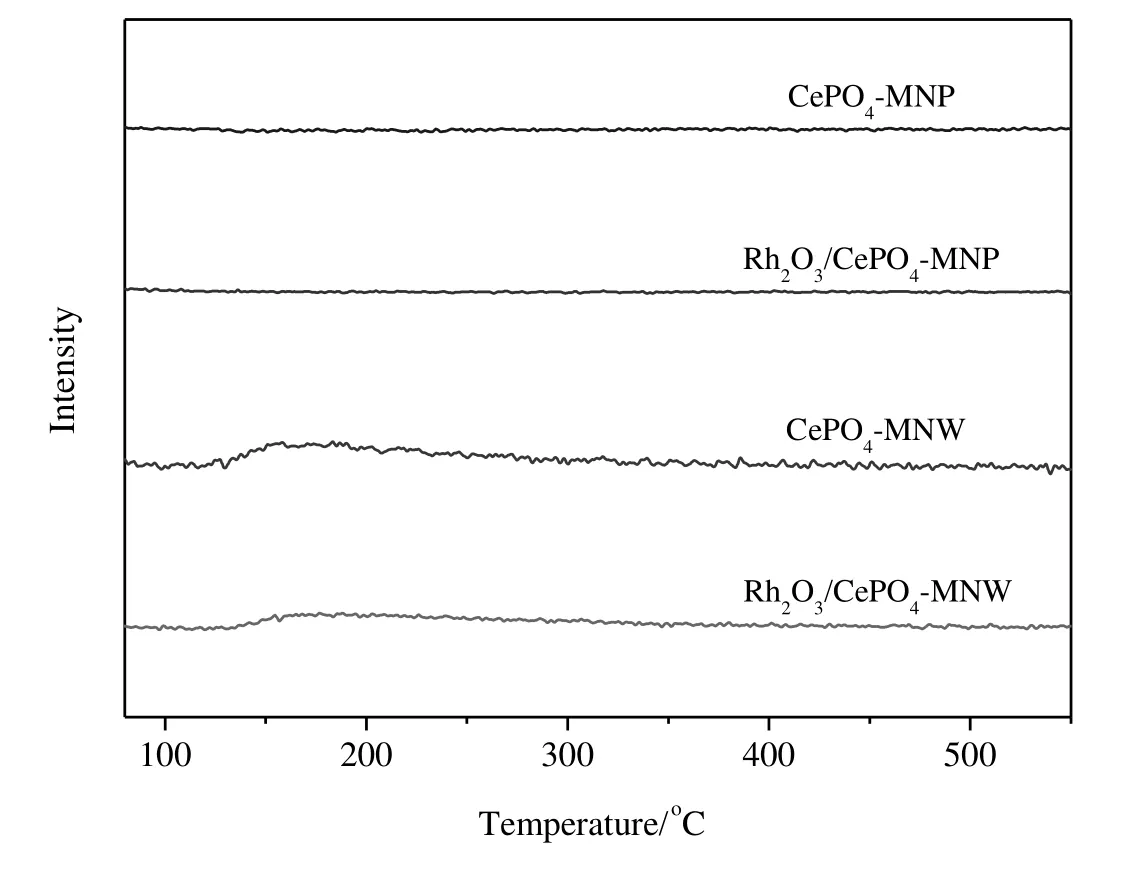
Fig.9.O2-TPD pro files of Rh2O3/CePO4 catalysts.
Fig.10 shows the H2-TPR pro files of Rh2O3/CePO4.These catalysts showa single peak corresponding to the reduction of Rh2O3to metallic Rh.The TPR main peaks of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are at 91 and 85°C,respectively.
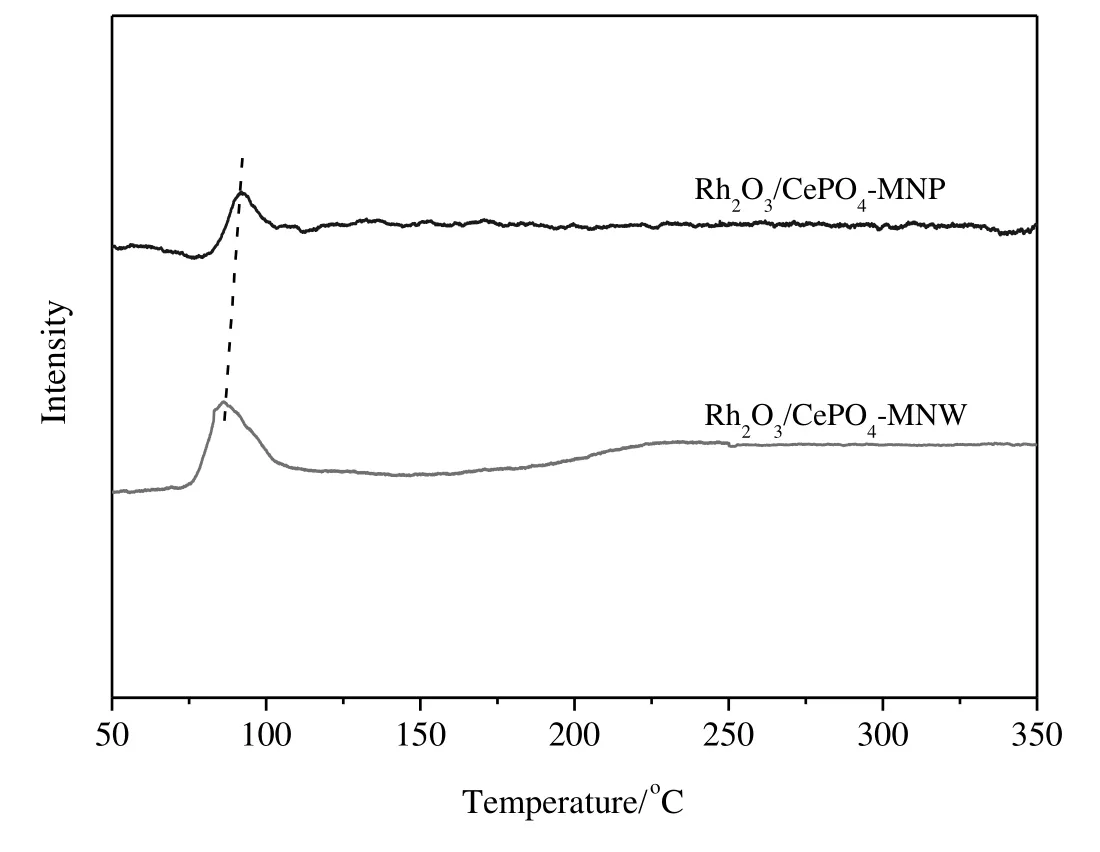
Fig.10.H2-TPR pro files of Rh2O3/CePO4 catalysts.
4.Discussion
In this work,monoclinic CePO4nanoparticles and nanowires were prepared by calcining hexagonal CePO4nanorods and CePO4nanowires at 900°C,respectively.TEM images(Fig.2)showthat CePO4-MNP is composed of large nanoparticles,whereas CePO4-MNW exhibits uniform nanowires.Both supports are monoclinic in nature,as confirmed by XRD data(Fig.1)and HRTEM images(Fig.2).These monoclinic CePO4materials were used to support Rh2O3.Rh2O3/CePO4-MNW is much more active than Rh2O3/CePO4-MNP in N2O decomposition(Fig.3)and CO oxidation(Fig.S6).
The Rh contents on CePO4-MNP and CePO4-MNW are measured by ICP-OES as 1.02 wt%and 0.99 wt%,all closed to 1 wt%.The Rh species is present as Rh2O3on both catalysts(Fig.S9).The average sizes of Rh2O3particles on CePO4-MNP and CePO4-MNW are(3.0±0.9)nm and(2.5±0.7)nm,respectively(Figs.S1 and S2).The specific surface areas of Rh2O3/CePO4-MNP and Rh2O3/CePO4-MNW are<1 m2·g−1and 16.6 m2·g−1,respectively.The size of Rh2O3particles seems to be related to the surface area,as supports with larger surface areas tend to disperse active species better.The size of Rh2O3species may Influence the activity of catalysts in N2O decomposition[64,65].
CO2-TPD data(Fig.8)show the basicity of supports and catalysts.It has been reported that the basic property of support is beneficial for N2O decomposition[10,12,66].Huang et al.[19]and Haber et al.[49]found some correlations among the basicity of support,the rhodium dispersion,and the catalytic activity.In the present work,the amount of basic sites of CePO4-MNW is 31 μmol·g−1,higher than that of CePO4-MNP(0 μmol·g−1),and Rh2O3nanoparticles disperse better on CePO4-MNW,as judged from the average sizes of Rh2O3nanoparticles on CePO4-MNW[(2.5±0.6)nm]and CePO4-MNP[(3.0±0.7)nm].This difference may be correlated to the difference in catalytic activity.
O2-TPD data(Fig.9)reveal that O2starts to desorb from Rh2O3/CePO4-MNW at 133°C.On the other hand,no O2desorbs from Rh2O3/CePO4-MNP,indicating that O2does not adsorb on Rh2O3/CePO4-MNP at 50°C(the adsorption temperature).For N2O decomposition,the desorption of O2from catalyst surface is considered as the ratedetermining step.If oxygen is not rapidly desorbed from the catalyst surface,it will be adsorbed on the active site,leading to the inhibition of N2O decomposition[10].Here the easy desorption or reversible adsorption of O2on both catalysts is consistent with the observation that the co-fed O2has a minor effect on the catalytic activity of both catalysts(Fig.5).The insignificant inhibiting effect of co-fed O2on Rh2O3/CePO4-MNP(Fig.6)versus Rh2O3/CePO4-MNW(Fig.7),as also seen in Fig.5,is consistent with the O2-TPD data(Fig.9).
H2-TPR data(Fig.10)show that Rh2O3on CePO4-MNW can be reduced at a lower temperature.The reducibility of supported metal oxide species was sometimes correlated to the catalytic performance in N2O decomposition[46,51,67–69].For instance,Zheng et al.[70]found that Ru Ox/TiO2is more active than Ru Ox/Al2O3,Ru Ox/SiO2,RuOx/CeO2,and RuOx/MgO in N2O decomposition because RuOxon TiO2can be reduced more easily.Imamura et al.[71]found that the relatively low reduction temperature of Rh Oxin RhOx/CeO2is correlated to its high catalytic activity in N2O decomposition.These observations are in line with our finding that Rh2O3/CePO4-MNW is more easily reduced than Rh2O3/CePO4-MNW and the former catalyst is more active than the latter.
The most active Rh2O3/CePO4-MNW is compared with other Rhbased catalysts.As shown in Table S1,the specific rate of Rh2O3/CePO4-MNW in N2O decomposition at 325 °Cis292 mmol·(g Rh)−1·h−1,higher than those of RhOx/LaPO4(26 mmol·(g·Rh)−1·h−1)[10],Rh2O3/γ-Al2O3(187 mmol·(g Rh)−1·h−1) [51],Rh2O3/SBA-15 (39 mmol·(g Rh)−1·h−1)[54],and Rh2O3/KIT-6(0 mmol·(g Rh)−1·h−1)[72],although lower than that of Rh2O3/mesoporous CoOx–Al2O3(324 mmol·(g Rh)−1·h−1)[56],Rh2O3/mesoporous Al2O3(306 mmol·(g Rh)−1·h−1)[56],and Rh2O3/CeO2(>1071 mmol·(g Rh)−1·h−1)[50].Further optimization of the catalyst is possible.As shown in Fig.S12,the pretreatment of Rh2O3/CePO4-MNW in 4%H2at 350°C for 3 h can enhance the catalytic activity to some extent,as the case with the pretreatment of Rh2O3/LaPO4[10]and Rh2O3/hydroxyapatite[10,19]in 4%H2.
As shown in Table S2,the specific rate of Rh2O3/CePO4-MNW in CO oxidation at 100 °C is 422 mmol·(g Rh)−1·h−1,higher than those of Rh2O3/mesoporous Al2O3(70 mmol·(g Rh)−1·h−1)[56],Rh2O3/mesoporous MnOx–Al2O3(119 mmol·(g Rh)−1·h−1)[56],and Rh2O3/γ-Al2O3(0 mmol·(g Rh)−1·h−1)[73],although lower than that of Rh2O3/LaPO4-nanowires(>515 mmol·(g Rh)−1·h−1)[11].It is difficult to compare the catalytic activities of Rh2O3/CePO4-MNW(422 mmol·(g Rh)−1·h−1),Rh/TiO2(>321 mmol·(g Rh)−1·h−1)[74],and Rh2O3/CeO2(>107 mmol·(g Rh)−1·h−1)[75]at 100 °C because the latter two catalysts show100%CO conversion below90°C.Thus,we chose alower reaction temperature for comparison.The specific rates of Rh2O3/CePO4-MNW,Rh2O3/CeO2[75],and Rh/TiO2[74]at 25°C are 43,28,and >321 mmol·(g Rh)−1·h−1,respectively(Table S3).
5.Conclusions
Monoclinic CePO4nanoparticles were prepared by calcining hexagonal CePO4nanorods(prepared by precipitation)at 900°C.Monoclinic CePO4nanowires were prepared by calcining hexagonal CePO4nanowires(prepared by hydrothermal synthesis at 150 °C)at 900 °C.CePO4-supported Rh2O3catalysts were tested in N2O decomposition and CO oxidation.Rh2O3/CePO4-MNW was found to be more active than Rh2O3/CePO4-MNP in both reactions.Rh2O3/CePO4-MNW has smaller Rh2O3particles,more basic sites,and low reduction temperature of Rh2O3species.The co-fed CO2had no effect on the activity of both catalysts.The co-fed O2only slightly inhibits catalytic activity.The co-fed H2O or O2+H2O can inhibit the activity,but the inhibiting effect is reversible on both catalysts.Although here we only studied N2O decomposition and CO oxidation,we believe that it would be interesting to study the applications of Rh2O3/CePO4catalysts in other reactions.
Supplementary Material
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.cjche.2017.02.007.
[1]W.F.Yan,S.Brown,Z.W.Pan,S.M.Mahurin,S.H.Overbury,S.Dai,Ultrastable gold nanocatalyst supported by nanosized non-oxide substrate,Angew.Chem.Int.Ed.45(2006)3614–3618.
[2]Z.Ma,H.F.Yin,S.H.Overbury,S.Dai,Metal phosphates as a newclass of supports for gold nanocatalysts,Catal.Lett.126(2008)20–30.
[3]Z.Ma,H.F.Yin,S.Dai,Influence of preparation methodson the performance of metal phosphate-supported gold catalysts in CO oxidation,Catal.Lett.138(2010)40–45.
[4]M.J.Li,Z.L.Wu,S.H.Overbury,CO oxidation on phosphate-supported Au catalysts:Effect of support reducibility on surface reactions,J.Catal.278(2011)133–142.
[5]H.Liu,Y.Lin,Z.Ma,Au/LaPO4nanowires:Synthesis,characterization,and catalytic CO oxidation,J.Taiwan Inst.Chem.Eng.62(2016)275–282.
[6]X.S.Qian,H.M.Qin,T.Meng,Y.Lin,Z.Ma,Metal phosphate-supported Pt catalysts for CO oxidation,Materials 7(2014)8105–8130.
[7]B.Pan,S.J.Luo,W.Y.Su,X.X.Wang,Photocatalytic CO2reduction with H2O over LaPO4nanorods deposited with Pt cocatalyst,Appl.Catal.B Environ.168–169(2015)458–464.
[8]H.Tamai,T.Ikeya,F.Nishiyama,H.Yasuda,K.Iida,S.Nojima,NO decomposition by ultra fine noble metals dispersed on the rare earth phosphate hollowparticles,J.Mater.Sci.35(2000)4945–4953.
[9]M.Machida,T.Eidome,S.Minami,H.P.Buwono,S.Hinokuma,Y.Nagao,Y.Nakahara,Tuning the electron density of Rh supported on metal phosphates for three-way catalysis,J.Phys.Chem.C 119(2015)11653–11661.
[10]Y.Lin,T.Meng,Z.Ma,Catalytic decomposition of N2O over RhOxsupported on metal phosphates,J.Ind.Eng.Chem.28(2015)138–146.
[11]H.Liu,Z.Ma,Effect of different LaPO4supports on the catalytic performance of Rh2O3/LaPO4in N2O decomposition and CO oxidation,J.Taiwan Inst.Chem.Eng.71(2017)373–380.
[12]Y.W.Cui,H.Liu,Y.Lin,Z.Ma,Metal phosphate-supported RuOxcatalysts for N2O decomposition,J.Taiwan Inst.Chem.Eng.67(2016)254–262.
[13]J.Huang,L.C.Wang,Y.M.Liu,Y.Cao,H.Y.He,K.N.Fan,Gold nanoparticles supported on hydroxylapatite as high performance catalysts for lowtemperature COoxidation,Appl.Catal.B Environ.101(2011)560–569.
[14]M.I.Domínguez,F.Romero-Sarria,M.A.Centeno,J.A.Odriozola,Gold/hydroxyapatite catalysts:Synthesis,characterization and catalytic activity to CO oxidation,Appl.Catal.B Environ.87(2009)245–251.
[15]K.F.Zhao,B.T.Qiao,J.H.Wang,Y.J.Zhang,T.Zhang,A highly active and sinteringresistant Au/FeOx-hydroxyapatite catalyst for CO oxidation,Chem.Commun.47(2011)1779–1781.
[16]N.Phonthammachai,Z.Y.Zhong,J.Guo,Y.F.Han,T.J.White,Synthesis of high performance hydroxyapatitie-gold catalysts for CO oxidation,Gold Bull.41(2008)42–50.
[17]H.Sun,F.Z.Su,J.Ni,Y.Cao,H.Y.He,K.N.Fan,Gold supported on hydroxyapatite as a versatile multifunctional catalyst for the direct tandem synthesis of imines and oximes,Angew.Chem.Int.Ed.Eng.48(2009)4390–4393.
[18]Y.Liu,H.Tsunoyama,T.Akita,S.Xie,T.Tsukuda,Aerobic oxidation of cyclohexane catalyzed by size-controlled Au clusters on hydroxyapatite:Size effect in the sub-2 nm regime,ACS Catal.1(2011)2–6.
[19]C.Y.Huang,Z.Ma,P.F.Xie,Y.H.Yue,W.M.Hua,Z.Gao,Hydroxy apatite-supported rhodium catalysts for N2O decomposition,J.Mol.Catal.A Chem.400(2015)90–94.
[20]C.Y.Huang,Y.X.Jiang,Z.Ma,P.F.Xie,Y.Lin,T.Meng,C.X.Miao,Y.H.Yue,W.M.Hua,Z.Gao,Correlation among preparation methods/conditions,physicochemical properties,and catalytic performance of Rh/hydroxyapatite catalysts in N2O decomposition,J.Mol.Catal.A Chem.420(2016)73–81.
[21]A.Venugopal,M.S.Scurrell,Hydroxyapatite as a novel support for gold and ruthenium catalysts:Behavior in the water gas shift reaction,Appl.Catal.A Gen.245(2003)137–147.
[22]J.W.Jaworski,D.Kim,K.Jung,S.Kim,J.H.Jung,J.O.Jeong,H.S.Jeon,B.K.Min,K.Y.Kwon,Surface modification of hydroxyapatite for hydrogen generation,J.Colloid Interface Sci.358(2011)598–603.
[23]Z.Opre,D.Ferri,F.Krumeich,T.Mallat,A.Baiker,Aerobic oxidation of alcohols by organically modified ruthenium hydroxyapatite,J.Catal.241(2006)287–295.
[24]Z.Opre,D.Ferri,F.Krumeich,T.Mallat,A.Baiker,Insight into the nature of active redox sites in Ru-containing hydroxyapatite by DRIFT spectroscopy,J.Catal.251(2007)48–58.
[25]C.Mondelli,D.Ferri,A.Baiker,Ruthenium at work in Ru-hydroxyapatite during the aerobic oxidation of benzyl alcohol:An in situ ATR-IR spectroscopy study,J.Catal.258(2008)170–176.
[26]Y.Takita,X.Qing,A.Takami,H.Nishiguchi,K.Nagaoka,Oxidative dehydrogenation of isobutane to isobutene III reaction mechanism over CePO4catalyst,Appl.Catal.A Gen.296(2005)63–69.
[27]Y.Takita,K.I.Sano,T.Muraya,H.Nishiguchi,N.Kawata,M.Ito,T.Akbay,T.Ishihara,Oxidative dehydrogenation of iso-butane to iso-butene II.Rare earth phosphate catalysts,Appl.Catal.A Gen.170(1998)21–23.
[28]C.Dume,W.F.HöÈlderich,Amination of 1-octanol,Appl.Catal.A Gen.183(1999)167–176.
[29]G.S.Devi,D.Giridhar,B.M.Reddy,Vapour phase O-alkylation of phenol over alkali promoted rare earth metal phosphates,J.Mol.Catal.A Chem.181(2002)173–178.
[30]H.Onoda,H.Nariai,A.Moriwaki,H.Maki,I.Motooka,Formation and catalytic characterization of various rare earth phosphates,J.Mater.Chem.12(2002)1754–1760.
[31]W.Y.Yao,Y.Liu,X.Q.Wang,X.L.Weng,H.Q.Wang,Z.B.Wu,The superior performance of sol–gel made Ce–O–P catalyst for selective catalytic reduction of NO with NH3,J.Phys.Chem.C 120(2016)221–229.
[32]F.Li,Y.B.Zhang,D.H.Xiao,D.Q.Wang,X.Q.Pan,X.G.Yang,Hydrothermal method prepared Ce–P–O catalyst for the selective catalytic reduction of NO with NH3in a broad temperature range,ChemCatChem 2(2010)1416–1419.
[33]Y.J.Zhang,J.H.Wang,T.Zhang,Novel Ca-doped CePO4supported ruthenium catalyst with superior catalytic performance for aerobic oxidation of alcohols,Chem.Commun.47(2011)5307.
[34]M.Itoh,M.Takehara,M.Saito,K.Machida,NOxreduction activity over phosphatesup ported platinum catalysts with hydrogen under oxygen-rich condition,IOP Conf.Ser.:Mater.Sci.Eng.18(2011)172007.
[35]F.Romero-Sarria,M.I.Domínguez,M.A.Centeno,J.A.Odriozola,CO oxidation at lowtemperature on Au/CePO4:Mechanistic aspects,Appl.Catal.B Environ.107(2011)268–273.
[36]S.Lucas,E.Champion,D.Bregiroux,D.Bernache-Assollant,F.Audubert,Rare earth phosphate powders RePO4·n H2O(Re=La,Ce or Y)—Part I.Synthesis and characterization,J.Solid State Chem.177(2004)1302–1311.
[37]X.L.Weng,R.J.Mei,M.P.Shi,Q.Y.Kong,Y.Liu,Z.B.Wu,CePO4catalyst for elemental mercury removal in simulated coal- fired fluegas,Energy Fuels29(2015)3359–3365.
[38]Y.P.Fang,A.W.Xu,R.Q.Song,H.X.Zhang,L.P.You,J.C.Yu,H.Q.Liu,Systematic synthesis and characterization of single-crystal lanthanide orthophosphate nanowires,J.Am.Chem.Soc.125(2003)16025–16034.
[39]Y.J.Zhang,H.M.Guan,Hydrothermal synthesisand characterization of hexagonal and monoclinic CePO4single-crystal nanowires,J.Cryst.Growth 256(2003)156–161.
[40]F.Y.Lu,Y.Q.Shen,X.Sun,Z.L.Dong,R.C.Ewing,J.Lian,Size dependence of radiationinduced amorphization and recrystallization of synthetic nanostructured CePO4monazite,Acta Mater.61(2013)2984–2992.
[41]D.Palma-Ramírez,M.A.Domínguez-Crespo,A.M.Torres-Huerta,H.Dorantes-Rosales,E.Ramírez-Meneses,E.Rodríguez,Microwave-assisted hydrothermal synthesis of CePO4nanostructures:Correlation between the structural and optical properties,J.Alloys Compd.643(2015)S209–S218.
[42]F.Kapteijn,J.Rodriguez-Mirasol,J.A.Moulijn,Heterogeneous catalytic decomposition of nitrous oxide,Appl.Catal.B Environ.9(1996)25–64.
[43]M.Konsolakis,Recent advances on nitrous oxide(N2O)decomposition over nonnoble-metal oxide catalysts:Catalytic performance,mechanistic considerations,and surface chemistry aspects,ACS Catal.5(2015)6397–6421.
[44]Z.M.Liu,F.He,L.L.Ma,S.Peng,Recent advances in catalytic decomposition of N2O on noble metal and metal oxide catalysts,Catal.Surv.Asia 20(2016)121–132.
[45]E.Kondratenko,V.Kondratenko,M.Santiago,J.Perezramirez,Mechanistic origin of the different activity of Rh-ZSM-5 and Fe-ZSM-5 in N2O decomposition,J.Catal.256(2008)248–258.
[46]H.Beyer,J.Emmerich,K.Chatziapostolou,K.Köhler,Decomposition of nitrous oxide by rhodium catalysts:Effect of rhodium particle size and metal oxide support,Appl.Catal.A Gen.391(2011)411–416.
[47]K.Yuzaki,T.Yarimizu,K.Aoyagi,S.Ito,K.Kunimori,Catalytic decomposition of N2O over supported Rh catalysts:Effects of supports and Rh dispersion,Catal.Today 45(1998)129–134.
[48]P.S.S.Reddy,N.Seshu Babu,N.Pasha,N.Lingaiah,P.S.Sai Prasad,Influence of microwave irradiation on catalytic decomposition of nitrous oxide over Rh/Al2O3catalyst,Catal.Commun.9(2008)2303–2307.
[49]J.Haber,M.Nattich,T.Machej,Alkali-metal promoted rhodium-on-alumina catalysts for nitrous oxide decomposition,Appl.Catal.B Environ.77(2008)278–283.
[50]A.Bueno-Lopez,I.Such-Basanez,C.S.M.D.Lecea,Stabilization of active Rh2O3species for catalytic decomposition of N2O on La-,Pr-doped CeO2,J.Catal.244(2006)102–112.
[51]S.Parres-Esclapez,M.J.Illán-Gómez,C.S.-M.de Lecea,A.Bueno-López,On the importance of the catalyst redox properties in the N2O decomposition over alumina and ceria supported Rh,Pd and Pt,Appl.Catal.B Environ.96(2010)370–378.
[52]M.Hussain,D.Fino,N.Russo,Development of modified KIT-6 and SBA-15-spherical supported Rh catalysts for N2O abatement:From powder to monolith supported catalysts,Chem.Eng.J.238(2014)198–205.
[53]J.M.Du,W.W.Kuang,H.L.Xu,W.Shen,D.Y.Zhao,The Influence of precursors on Rh/SBA-15 catalysts for N2O decomposition,Appl.Catal.B Environ.84(2008)490–496.
[54]L.Chmielarz,P.Kuśtrowski,M.Drozdek,M.Rutkowska,R.Dziembaj,M.Michalik,P.Cool,E.F.Vansant,SBA-15 mesoporous silicamodified with rhodium by MDDmethod and its catalytic role for N2O decomposition reaction,J.Porous.Mater.18(2010)483–491.
[55]L.Kuboňová,D.Fridrichová,A.Wach,P.Kuśtrowski,L.Obalová,P.Cool,Catalytic activity of rhodium grafted on ordered mesoporous silica materials modified with aluminum in N2O decomposition,Catal.Today 257(2015)51–58.
[56]H.Liu,Y.Lin,Z.Ma,Rh2O3/mesoporous MOx–Al2O3(M=Mn,Fe,Co,Ni,Cu,Ba)catalysts:Synthesis,characterization,and catalytic applications,Chin.J.Catal.37(2016)73–82.
[57]T.Meng,N.Ren,Z.Ma,Silicalite-1@Cu-ZSM-5 core-shell catalyst for N2O decomposition,J.Mol.Catal.A Chem.404–405(2015)233–239.
[58]M.Cao,C.Hu,Q.Wu,C.Guo,Y.Qi,E.Wang,Controlled synthesis of LaPO4and CePO4nanorods/nanowires,Nanotechnology 16(2005)282–286.
[59]L.Li,S.F.Niu,Y.Qu,Q.Zhang,H.Li,Y.S.Li,W.R.Zhao,J.L.Shi,One-pot synthesis of uniform mesoporous rhodium oxide/alumina hybrid as high sensitivity and lowpower consumption methane catalytic combustion micro-sensor,J.Mater.Chem.22(2012)9263–9267.
[60]M.Machida,S.Minami,S.Hinokuma,H.Yoshida,Y.Nagao,T.Sato,Y.Nakahara,Unusual redox behavior of Rh/AlPO4and its impact on three-way catalysis,J.Phys.Chem.C 119(2015)373–380.
[61]L.M.Qiu,F.Liu,L.Z.Zhao,Y.Ma,J.N.Yao,Comparative XPS study of surface reduction for nanocrystalline and microcrystalline ceria powder,Appl.Surf.Sci.252(2006)4931–4935.
[62]E.Bêche,P.Charvin,D.Perarnau,S.Abanades,G.Flamant,Ce 3d XPS investigation of cerium oxides and mixed cerium oxide(CexTiyOz),Surf.Interface Anal.40(2008)264–267.
[63]K.F.Zhao,H.L.Tang,B.T.Qiao,L.Li,J.H.Wang,High activity of Au/γ-Fe2O3for CO oxidation:Effect of support crystal phase in catalyst design,ACS Catal.5(2015)3528–3539.
[64]M.Piumetti,M.Hussain,D.Fino,N.Russo,Mesoporous silica supported Rh catalysts for high concentration N2Odecomposition,Appl.Catal.BEnviron.165(2015)158–168.
[65]S.Parres-Esclapez,F.E.López-Suárez,A.Bueno-López,M.J.Illán-Gómez,B.Ura,J.Trawczynski,Rh–Sr/Al2O3catalyst for N2O decomposition in the presence of O2,Top.Catal.52(2009)1832–1836.
[66]L.Obalová,K.Karásková,A.Wach,P.Kustrowski,K.Mamulová-Kutláková,S.Michalik,K.Jirátová,Alkali metals as promoters in Co–Mn–Al mixed oxide for N2O decomposition,Appl.Catal.A Gen.462–463(2013)227–235.
[67]L.Obalová,K.Jirátová,F.Kovanda,K.Pacultová,Z.Lacný,Z.Mikulová,Catalytic decomp osition of nitrous oxide over catalysts prepared from Co/Mg-Mn/Al hydrotalcite-like compounds,Appl.Catal.B Environ.60(2005)289–297.
[68]L.Xue,H.He,C.Liu,C.B.Zhang,B.Zhang,Promotion effects and mechanism of alkali metals and alkaline earth metals on cobalt–cerium composite oxide catalysts for N2O decomposition,Environ.Sci.Technol.43(2009)890–895.
[69]Z.Ma,Y.Ren,Y.Lu,P.G.Bruce,Catalytic decomposition of N2O on ordered crystalline metal oxides,J.Nanosci.Nanotechnol.13(2013)5093–5103.
[70]J.Zheng,S.Meyer,K.Köhler,Abatement of nitrous oxide by ruthenium catalysts:Influence of the support,Appl.Catal.A Gen.505(2015)44–51.
[71]S.Imamura,J.I.Tadani,Y.Saito,Y.Okamoto,H.Jindai,C.Kaito,Decomposition of N2O on Rh-loaded Pr/Ce composite oxides,Appl.Catal.A Gen.201(2000)121–127.
[72]M.Hussain,P.Akhter,D.Fino,N.Russo,Modified KIT-6 and SBA-15-sphericalsupported metal catalysts for N2O decomposition,J.Environ.Chem.Eng.1(2013)164–174.
[73]Y.P.Cai,H.G.Stenger Jr.,C.E.Lyman,Catalytic CO oxidation over Pt-Rh/γ-Al2O3catalysts,J.Catal.161(1996)123–131.
[74]H.Guan,J.Lin,B.Qiao,X.Yang,L.Li,S.Miao,J.Y.Liu,A.Q.Wang,X.D.Wang,T.Zhang,Catalytically active Rh sub-nanoclusters on TiO2for CO oxidation at cryogenic temperatures,Angew.Chem.Int.Ed.55(2016)2820–2824.
[75]V.R.Pérez,Á.V.M.Beltrán,Q.G.He,Q.Wang,C.S.M.D.Lecea,B.A.López,Preparation of ceria-supported rhodium oxide sub-nanoparticles with improved catalytic activity for CO oxidation,Catal.Commun.33(2013)47–50.
猜你喜欢
疯狂英语·初中天地(2022年1期)2022-07-07
国企管理(2022年3期)2022-05-17
小学生优秀作文(低年级)(2022年3期)2022-03-29
包头职业技术学院学报(2021年3期)2021-11-19
当代工人(2019年11期)2019-07-10
儿童故事画报(2017年3期)2017-05-26
大江南北(2016年6期)2016-11-21
湖北开放大学学报(2016年5期)2016-06-05
中国教育技术装备(2015年6期)2015-03-01
小说月刊(2014年11期)2014-04-18
 Chinese Journal of Chemical Engineering2018年1期
Chinese Journal of Chemical Engineering2018年1期
- Chinese Journal of Chemical Engineering的其它文章
- Membrane materials in the pervaporation separation of aromatic/aliphatic hydrocarbon mixtures—A review☆
- Cultivation of microalgae for biodiesel production:A reviewon upstream and downstream processing☆
- Numerical study and acceleration of LBM-RANS simulation of turbulent flow☆
- GPU-based discrete element simulation on flow stability of flat-bottomed hopper☆
- Tuning sol size to optimize organosilica membranes for gas separation☆
- Oil–water pre-separation with a novel axial hydrocyclone☆