
三环己基锡2-萘甲酸酯配合物的合成、晶体结构及体外抗癌活性研究
2018-05-24 06:25成英杰朱小明王一波黄修辉何哲超庾江喜张复兴邝代治
山东化工 2018年9期
成英杰,朱小明,2,3*,王一波,黄修辉,刘 莺,何哲超,庾江喜,2,3,张复兴,2,3,邝代治,2,3
(1.衡阳师范学院 化学与材料科学系,湖南 衡阳 421008;2.功能金属有机化合物湖南省重点实验室,湖南 衡阳 421008;3.功能金属有机材料湖南省普通高等学校重点实验室,湖南 衡阳 421008)
有机锡羧酸酯由于其结构和性能的多样性,尤其是其具有良好的抗癌和杀菌生物活性,成为人们研究热点[1-2]。2-萘甲酸用于植物生长调节剂,及较大的共轭平面,与毒性较大的小位阻烃基锡如三甲基锡[3]和二烃基锡[4]结合配位已有文献报道,而与大位阻烃基锡结合还未见报道。因此本文选择空间位阻较大的、毒性较小的三环己基氢氧化锡与2-萘甲酸反应,合成了标题配合物,对其进行了元素分析、红外光谱及核磁共振(1H,13C,119Sn)表征,通过X-射线晶体衍射仪测定了其晶体结构,并对其进行了热稳定性和体外抗癌活性研究。
1 实验部分
1.1 试剂与仪器
Avance III HD 500MHz全数字化超导核磁共振谱仪(瑞士布鲁克公司),Bruker SMART APEX II CCD单晶衍射仪(德国Bruker公司),日本岛津IRPrestige-21红外光谱仪(4000 cm-1~400 cm-1,KBr)测定,TGA Q50热重分析仪(美国TGA仪器公司),PE-2400(Ⅱ)型元素分析仪(美国PE公司)。
三环己基氢氧化锡为化学纯,其余试剂均为分析纯。人癌细胞株均取自美国模式培养物集存库,含10%胎牛血清的RPMI-1640培养基购自美国Gibico公司,胰蛋白酶购自甘肃金盛生化制药有限公司。
1.2 配合物的合成
在圆底烧瓶中按顺序依次加入萘甲酸0.172 g(1 mmol)、三环己基氢氧化锡0.385 g(1 mmol)、溶剂无水甲醇20 mL,搅拌回流8 h;冷却,过滤,在室温的条件下,溶剂自然挥发结晶,得无色透明晶体0.388 g,产率68%。m.p.: 205 ℃~206 ℃。元素分析:实测值(计算值,%):C,62.91(63.06),H,7.81(7.76)。IR(KBr,v/cm-1): 2918,2845(s,v(C-H)),1639(m,vas(COO-)),1329(m,vs(COO-)),605(m,v(Sn-C)),416(w,v(Sn-O))。1H NMR(CDCl3,500 MHz),δ 8.63(s,1H),8.11(dd,J=8.5 Hz,J=1.6 Hz,1H),7.95(d,J=10.7 Hz,1H),7.88~7.85(m,2H),7.57~7.49(m,2H),3.49(s,3H),2.08~1.32(m,33H),1.04(s,1H)。13C NMR(CDCl3,125 MHz),δ 171.46,135.15,132.65,131.19,129.62,129.28,127.68,127.60,126.47,126.23,50.91,33.94,31.17,28.96,26.94。119Sn NMR(CDCl3,186 MHz),δ (ppm): 16.09。
1.3 配合物的体外抗癌活性测试
采用四氮唑盐还原法(MTT法)测定配合物对人癌细胞NCI-H460、MCF-7和HepG2增殖的抑制活性,实验数据应用Graph Pad Prism 5.0统计软件分析,通过存活率百分比数据相对于药物浓度的非线性回归分析(曲线拟合),用S形剂量响应(变量)方程确定IC50值[5]。
1.4 配合物晶体结构的测定
选取尺寸合适的配合物待测晶体,在单晶衍射仪上,采用经石墨单色化的MoKα射线,于296(2) K,以φ~ω扫描方式收集数据。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,部分非氢原子坐标在随后的数轮差值Fourier合成中陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对氢原子和非氢原子分别采用各向同性和各向异性热参数进行全矩阵最小二乘法修正。全部结构分析计算工作采用SHEXTL-97程序系统完成。
2 结果与讨论
2.1 谱学分析
在配合物红外图谱中,2-萘甲酸的C=O伸缩振动尖锐吸收峰1703 cm-1和O-H弥散的伸缩振动吸收峰2533~3053 cm-1均消失,并在416 cm-1处出现了新的Sn-O键的伸缩振动吸收峰,说明在2-萘甲酸中的羧酸基团已脱去氢质子与Sn发生了配位[5]。配合物在1639 cm-1和1354 cm-1出现了新的尖锐吸收峰,分别为萘甲酸根上COO-基团的反对称伸缩振动和对称伸缩振动吸收峰,其峰值差Δν为285 cm-1,表明萘甲酸的羧酸基团脱去氢质子以羧基氧原子与Sn发生了单齿配位[6]。
2.2 晶体结构分析
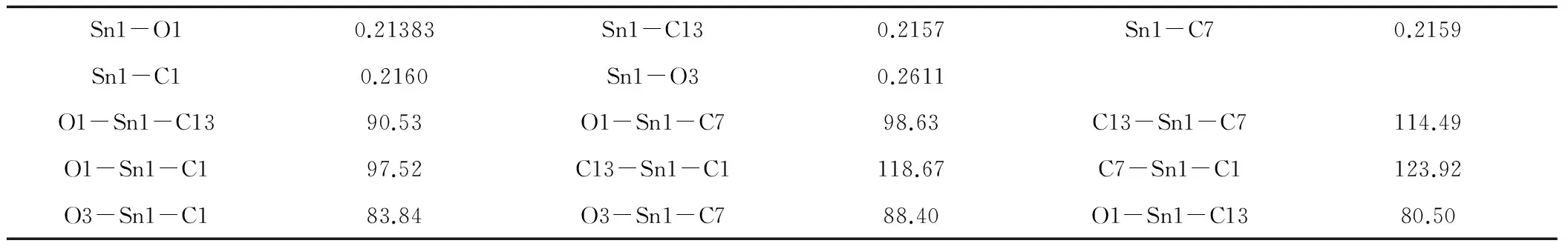
表1 配合物的主要键长(nm)和键角(°)
配合物晶体结构见图1,配合物的主要键长与键角见表1。晶体结构分析表明,在配合物中,中心锡原子分别与3个环己基的3个碳原子、1个羧基氧原子和1个甲醇氧原子配位形成五配位三角双锥构型。位于赤道位置上的C7-Sn1-C1、C13-Sn1-C1、C13-Sn1-C7的键角分别为123.92°、118.67°、114.49°,总键角之和为357.08°,接近360°,表明C1、C13、C7与Sn1几乎在一个平面上。轴向位置O1-Sn1-O3的键角为170.35°与180°偏离较大。从键长和键角数据表明配合物是以锡原子为中心形成了扭曲的三角双锥构型。
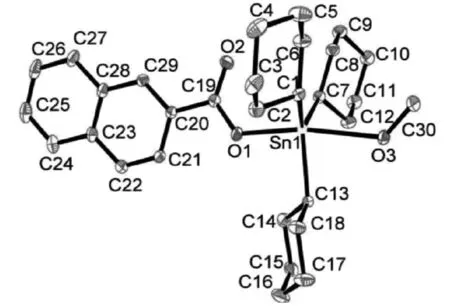
图1 配合物的分子结构图(椭球率15%,氢原子被省略)
2.3 配合物的热稳定性研究

图2 配合物的TG曲线
在氮气保护下,加热速度为10 ℃·min-1,在50~800 ℃范围对配合物进行了实验,测得配合物的热重分析曲线如图2所示,配合物的最大失重温区为240~500 ℃,失重区归属于三环己基和2-萘甲酸基基团的离去,失重率为74.07%。假定残余物对应的是SnO2,理论计算值分别为23.58%,实测值与计算值基本吻合。结果表明,配合物在75 ℃以下都可以稳定存在。
2.4 体外抗肿瘤活性
对配合物进行体外抗癌活性测试,结果表明:配合物对人肺癌细胞NCI-H460、人乳腺癌细胞MCF-7和人肝癌细胞HepG2增殖均有较高的抑制活性,其IC50值分别为0.30、0.34和0.35 μmol·L-1。
3 结论
合成了三环己基锡2-萘甲酸酯配合物,配合物中锡原子为扭曲三角双锥构型。体外抗癌活性测试表明,配合物具有较好的抗癌活性,可望作为广谱抗癌的候选药物。
参考文献
[1]Du D,Jiang Z,Liu C,et al.Macrocyclic organotin(IV) carboxylates based on benzenedicarboxylic acid derivatives: syntheses,crystal structures and antitumor activities[J].J Organomet Chem,2011,696(13):2549-2558.
[2]周昕鑫,赵佳伟,黄 敏,等.梯形四核有机锡氧簇合物[(μ-O)(μ-OMe)(6-BrC9H4O2CO2)(Bn2Sn)2]2的合成及晶体结构研究[J].山东化工,2017,46(10):1-3,6.
[3]Begley M J,Sowerby D B,Kapoor P,et al.Trimethyltin(IV) naphthoates: X-ray structure of the 2-isomer[J].Polyhedron,1995,14(13-14):1937-1941.
[4]Kappor R,Gupta A,Kapoor P,et al.Solution and solid state molecular structures of dialkyltin(IV) β-naphthoates: [{R2Sn(OCOC10H7)}2O]2,R2Sn(OCOC10H7)2and (CH3)4N+[R2Sn(OCOC10H7)3]-(R=n-C4H9and CH3).X-ray crystal structures of [{(n-C4H9)2Sn(OCOC10H7)}2O]2and [(n-C4H9)2Sn(OCOC10H7)2][J].Main Group Met Chem,2002,25(10):635-642.
[5]朱小明,邝代治,冯泳兰,等.双[三(2-甲基-2-苯基)丙基锡]二元酸酯(CH2)n[CO2Sn(CH2CMe2Ph)3]2(n=5,6)的合成、结构、热稳定性及生物活性[J].无机化学学报,2015,31(7):1373-1379.
[6]Ashfaq M,Khan M I,Baloch M K,et al.Biologically potent organotin(IV) complexes of 2-maleimidoacetic acid[J].J Organomet Chem,2004,689(1):238-245.
猜你喜欢
高中数理化(2022年16期)2022-09-14
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
中学物理·高中(2016年8期)2016-08-08
衡阳师范学院学报(2016年3期)2016-07-10
原子能科学技术(2015年4期)2015-05-25
火炸药学报(2014年3期)2014-03-20
郑州大学学报(理学版)(2013年2期)2013-03-11
通化师范学院学报(2012年2期)2012-01-11
