
丹酚酸A对脂多糖诱导人肺微血管内皮细胞线粒体自噬和细胞损伤的影响
2018-05-11 00:02:40李伟文蓝秀黎媛孙蕾吕祝庆
温州医科大学学报 2018年4期
李伟文,蓝秀,黎媛,孙蕾,吕祝庆
(温州医科大学附属第五医院 呼吸内科,浙江 丽水 323000)
人肺微血管内皮细胞(human pulmonary microvascular endothelial cells,HPMVECs)是构成肺泡-毛细血管屏障的重要成分。在肺炎症性疾病中,内毒素诱导的HPMVECs损伤是导致肺泡-毛细血管屏障破坏,最终导致急性肺损伤,甚至导致急性呼吸窘迫综合征的重要原因之一[1-2],预防和治疗HPMVECs损伤是降低患者病死率的重要手段[2-3]。丹酚酸A(salvi-anolic acid A,Sal A)是从丹参根茎中提取的重要活性成分,具有清除氧自由基、降低细胞内氧化应激、减少细胞损伤的作用[4-5]。本研究以脂多糖(lipopolysaccharides,LPS)作为诱导剂,构建HPMVECs损伤模型,观察Sal A对LPS诱导的HPMVECs损伤的保护作用,探讨其可能的作用机制。
1 材料和方法
1.1 材料 HPMVECs购于美国ScienCell公司,Sal A、MTT、RPMI 1640培养基购于美国Sigma公司,10%胎牛血清购于美国Gibco公司,ROS检测试剂盒和JC1试剂盒购自上海碧云天生物技术有限公司,线粒体蛋白提取试剂盒购于上海贝博生物科技有限公司,Beclin1、LC3 II/I、PINK1、Parkin和Tubulin单克隆抗体购于美国CST公司。
1.2 方法
1.2.1 细胞培养:HPMVECs用含链霉素、氯霉素和10%胎牛血清的RPMI 1640培养基传代,培养于37 ℃、5% CO2、饱和湿度的细胞培养箱中。细胞融合达80%以上时传代,取第4~第8代细胞进行实验。
1.2.2 细胞增殖检测:按照文献[6]方法,以细胞密度为2×104/mL接种于96孔板,每孔200 μL,次日换无血清培养基。用0.5、2.5、5、10、25、50 μmol/L Sal A处理48 h;分为4组:对照组、Sal A组、LPS组、LPS+Sal A组,Sal A组用终浓度为50 μmol/L的Sal A处理,LPS组用终浓度为10 μg/mL LPS处理,LPS+Sal A组用10 μg/mL的LPS和50 μmol/L的Sal A共培养,对照组不加药,并设置空白对照为调零孔,每组设置6个复孔,继续培养24 h、48 h和72 h。经上述处理后,每孔加入20 μL MTT试剂,加入二甲基亚砜后,用酶联免疫检测仪在492 nm波长处测各孔的吸光度值(A值)。细胞相对存活率=(受试组A值-空白对照A值)/(对照组A值-空白对照A值)×100%。
1.2.3 ROS相对含量和线粒体膜电位检测:按上述分组,继续培养48 h后,按照ROS试剂盒检测方法,用终浓度为10 μmol/L的DCFH-CA重悬细胞,置入37 ℃细胞培养箱内孵育20 min,以无血清细胞培养液洗涤3次后,用流式细胞技术分析细胞内ROS含量改变。按照JC1试剂盒说明书操作方法收集1×106个细胞,用JC1染色缓冲液洗涤2次,加入10 μg/mL的JC1试剂,置入37 ℃细胞培养箱内孵育20 min,用流式细胞仪检测红色荧光强度改变。以对照物为标准,计算各组ROS和JC1的相对含量。
1.2.4 蛋白提取及浓度测定:按上述分组继续培养48 h后,弃上清,用预冷PBS漂洗2次,加入120~150 μL RIPA裂解液冰上裂解10~15 min,4 ℃离心机12000 r/min离心15 min,取上清,即为细胞总蛋白。根据线粒体提取试剂盒说明书,将各组细胞培养48 h后,收集细胞,加入200 μL试剂A,冰上静置10 min,匀浆后,离心,取上清液,11000×g离心后,弃上清,加入试剂B,再次离心后,取沉淀物,加入100 μL的裂解液,收集线粒体蛋白。采用BCA法检测细胞总蛋白和线粒体蛋白浓度。
1.2.5 免疫印迹法检测细胞及线粒体自噬蛋白改变:取20 μg细胞总蛋白或者线粒体蛋白4%~12%聚丙烯酰胺梯度凝胶电泳分离,电转至PVDF膜,3%脱脂牛奶室温封闭30~60 min。加入一抗(Beclin1、LC3 II/I、PINK1、Parkin和Tubulin),4 ℃孵育过夜;PBST缓冲液洗涤3次,每次8~10 min,加入HRP标记二抗,室温孵育1~2 h,PBST缓冲液洗膜3次,每次8~10 min,用ECL试剂发光、显影和定影,Image J软件进行蛋白显带颜色深浅和面积计算,以Tubulin为内参。上述实验重复3次。
1.3 统计学处理方法 采用SPSS22.0统计软件进行分析。计量资料以±s表示,采用单因素方差分析和重复测量资料方差分析。P<0.05为差异有统计学意义。
2 结果
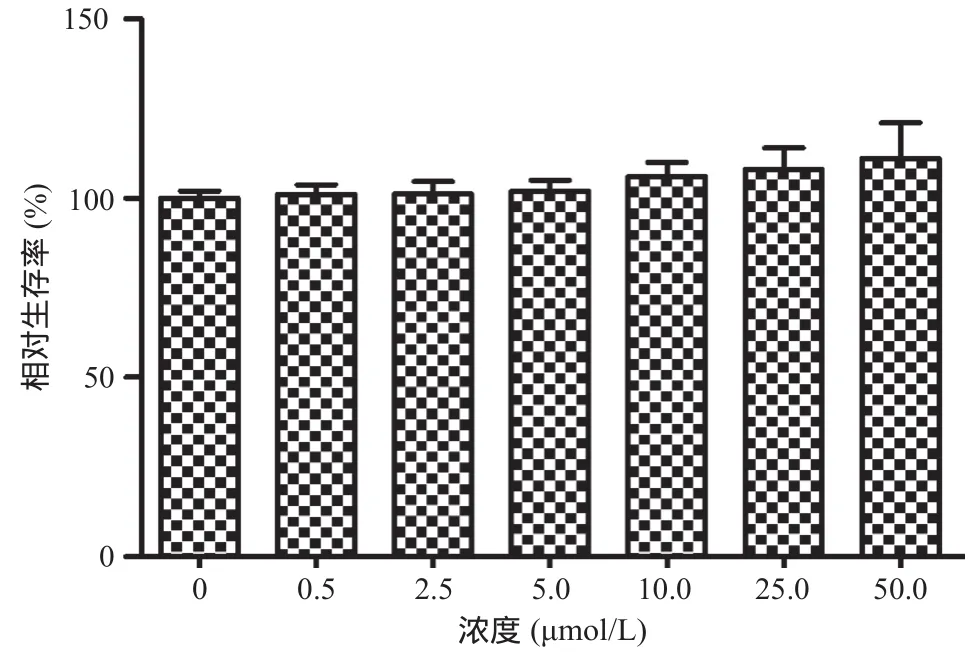
图1 Sal A对HPMVECs相对存活率的影响
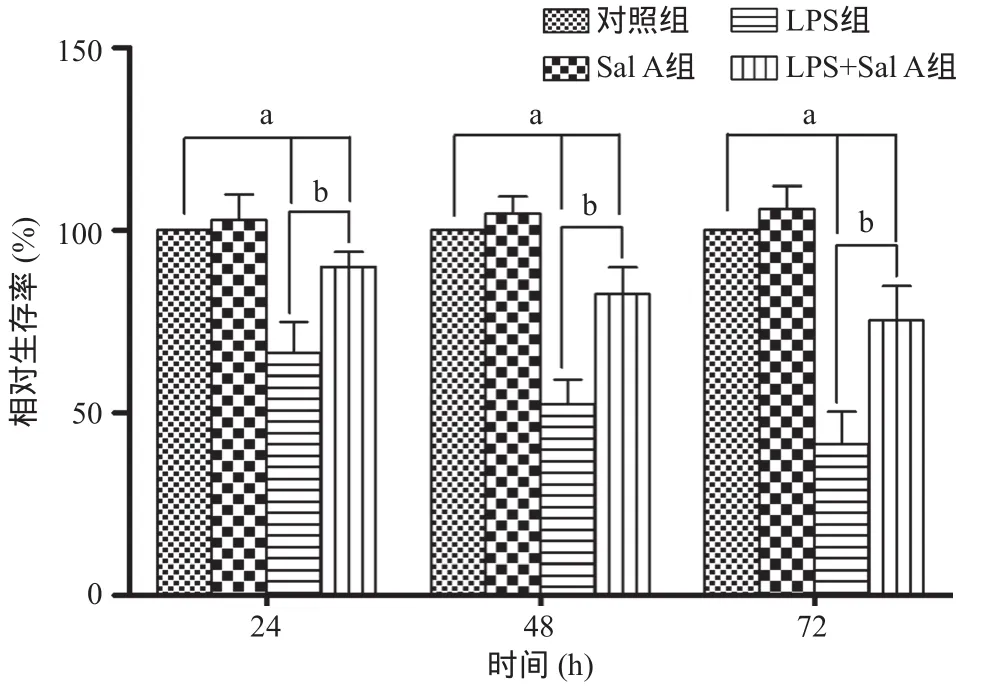
图2 Sal A对LPS诱导HPMVECs损伤的影响
2.1 Sal A抑制LPS诱导的细胞损伤作用 图1显示以终浓度为0.5、2.5、5、10、25、50 μmol/L的Sal A处理HPMVECs 48 h后,细胞增殖无显著抑制,与对照组比差异无统计学意义(P>0.05)。图2显示,50 μmol/L Sal A处理24 h、48 h和72 h后,细胞相对存活率无明显降低,与对照组比差异无统计学意义(P>0.05)。10 μg/mL LPS处理24 h、48 h和72 h后,细胞相对存活率降低;与LPS组比,LPS+Sal A组24 h、48 h和72 h时细胞相对存活率显著增高(P<0.05),但低于对照组(P<0.05)。
2.2 Sal A对LPS诱导HPMVECs的ROS含量和线粒体膜电位的影响 ROS活性检测结果显示:LPS组较对照组显著增高(P<0.05),Sal A组与对照组比较差异无统计学意义(P>0.05),LPS+Sal A组与对照组比较差异无统计学意义(P>0.05),但低于LPS组(P<0.05)。线粒体膜电位检测结果显示:LPS组较对照组显著降低(P<0.05),Sal A组与对照组比较差异无统计学意义(P>0.05),LPS+Sal A组显著低于对照组,高于LPS组(P<0.05)。见表1。
2.3 Sal A调节LPS诱导线粒体自噬水平 细胞自噬蛋白检测结果:Sal A组LC3- II/I、Beclin1、PINK1和Parkin相对表达量与对照组比差异无统计学意义(P>0.05),LPS组LC3- II/I、Beclin1、PINK1和Parkin蛋白相对表达量较对照组均显著增高(P<0.05),LPS+Sal A组LC3- II/I、Beclin1、PINK1和Parkin蛋白相对表达量与对照组比差异无统计学意义(P>0.05),但低于LPS组(P<0.05)。见表2。线粒体自噬蛋白检测结果:Sal A组PINK1和Parkin相对表达量与对照组比差异无统计学意义(P>0.05),LPS组PINK1和Parkin蛋白相对表达量与对照组比均显著增高(P<0.05);LPS+Sal A组PINK1和Parkin蛋白相对表达量与对照组比差异无统计学意义(P>0.05),但低于LPS组(P<0.05)。见表3。
表1 各组ROS含量和线粒体膜电位比较(n=3,±s)
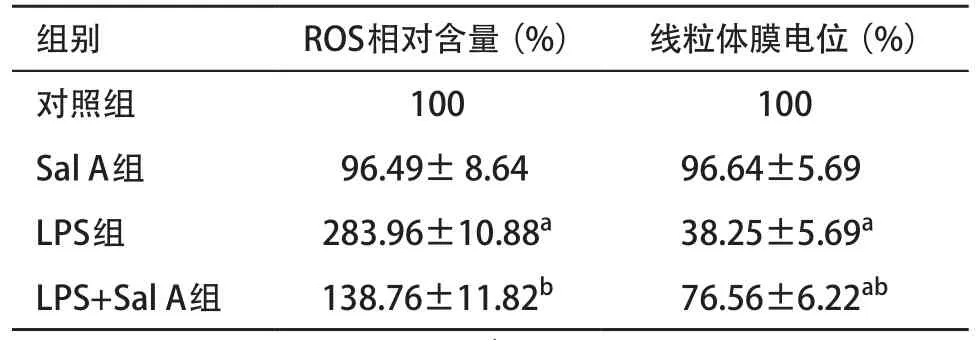
表1 各组ROS含量和线粒体膜电位比较(n=3,±s)
与对照组比:aP<0.05;与LPS组比:bP<0.05
3 讨论
LPS诱导的HPMVECs炎性损伤模型是目前常用的体外模拟肺泡-毛细血管屏障损害模型[2]。本研究以该模型作为工具,分析Sal A对LPS诱导细胞炎性损伤的保护作用,结果显示,10 μg/mL LPS处理HPMVECs 24 h后,细胞相对存活率显著降低,且随着时间的延长,细胞相对存活率逐渐下降,说明10 μg/mL LPS成功构建细胞损伤模型;终浓度为0.5、2.5、5、10、25、50 μmol/L的Sal A对HPMVECs无显著抑制作用。以终浓度为50 μmol/L的Sal A与LPS共同处理HPMVECs后,细胞相对存活率较LPS组显著增加,说明Sal A具有抑制LPS诱导HPMVECs损伤作用。
线粒体是机体能量代谢的中心,是细胞内ROS的主要来源之一[7]。适量的ROS有利于细胞内信号传导、基因表达的启动,促进增殖和分化,但过量ROS诱发氧化应激导致线粒体功能异常、细胞损伤、凋亡[8-9]。有学者认为线粒体损伤初期通过正反馈调节途径,诱发ROS爆发性增高,导致线粒体膜电位改变;靶向性降低细胞内ROS,可显著抑制线粒体功能异常[9-10]。本研究中,10 μg/mL LPS处理HPMVECs 48 h后,细胞内ROS含量显著增高,线粒体膜电位显著降低,提示LPS诱导线粒体损伤。50 μmol/L的Sal A显著抑制LPS诱导的细胞内ROS含量增高和线粒体膜电位下降,与文献[5,11]报道一致。
表2 各组细胞自噬相关蛋白表达比较(n=3,±s)
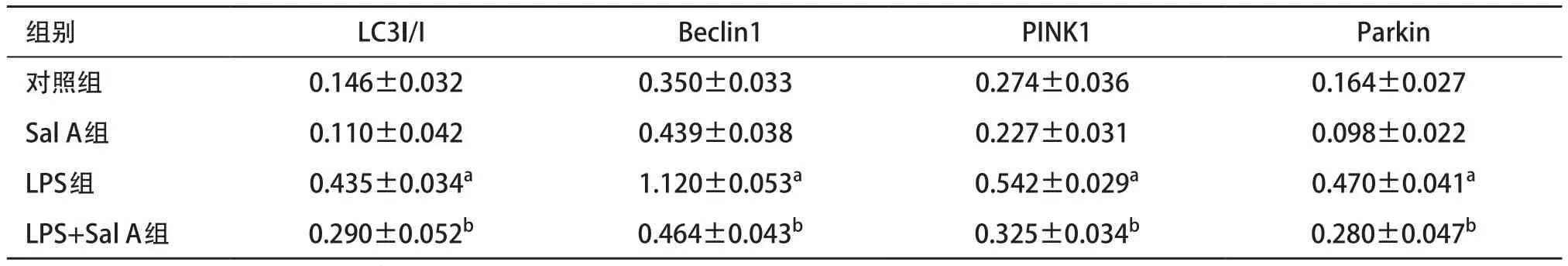
表2 各组细胞自噬相关蛋白表达比较(n=3,±s)
与对照组比:aP<0.05;与LPS组比:bP<0.05
表3 各组线粒体PINK1和Parkin蛋白表达比较(n=3,±s)
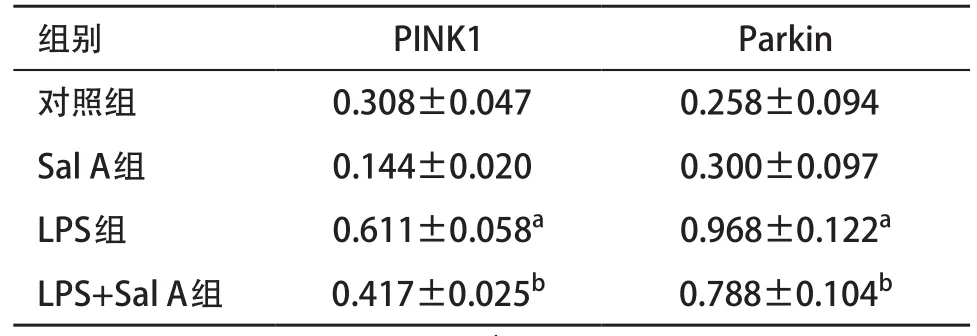
表3 各组线粒体PINK1和Parkin蛋白表达比较(n=3,±s)
与对照组比:aP<0.05;与LPS组比:bP<0.05
线粒体自噬是细胞通过自噬清除损伤线粒体的过程,能够介导氧化应激引起的细胞损伤[9,12]。ROS增加细胞内PINK1表达,并使PINK1募集Parkin于线粒体上,促进自噬体吞噬损伤的线粒体[13];而氧化清除剂N-乙酰半胱氨酸通过清除细胞内ROS,抑制LC3- II/LC3-I和PINK1表达[14]。FENG等[15]发现抑制过氧硝酸盐诱导的线粒体自噬能抑制脑缺血再灌注损伤。TANG等[16]发现人参皂苷通过调控线粒体自噬平衡,抑制糖氧缺乏诱导的神经胶质细胞损伤。郑丽云等[17]研究发现红景天苷能够通过线粒体自噬抑制糖氧缺乏星形胶质细胞损伤。本研究结果显示,与对照组相比,LPS组LC3- II/I、Beclin1、PINK1、Parkin表达显著增高,说明LPS处理后细胞内自噬水平增加,而用LPS和Sal A共培养后,LC3- II/I、Beclin1、PINK1、Parkin表达降低,说明Sal A拮抗LPS诱导线粒体自噬。进一步提取线粒体蛋白,证实Sal A显著降低LPS诱导的PINK1、Parkin表达。
综上所述,本研究证实Sal A对LPS诱导的HPMVECs具有保护作用,但由于实验过程中未应用阳性对照,仅反映了Sal A降低了LPS诱导的ROS含量,稳定了线粒体膜电位和降低了线粒体自噬水平,仍无法证实三者及其相互关系在Sal A抑制LPS诱导HPMVECs损伤中的作用机制,仍需进一步研究。
参考文献:
[1] BERNARD G. Acute lung failure-our evolving understanding of ARDS[J]. N Engl J Med, 2017, 377(6): 507-509.
[2] ZOU Y, BAO S, WANG F, et al. FN14 blockade on pulmonary microvascular endothelial cells improves the outcome of sepsis-induced acute lung injury[J]. Shock, 2018, 49(2):213-220.
[3] STANDIFORD T J, WARD P A. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome[J].Transl Res, 2016, 167(1): 183-191.
[4] FAN H Y, YANG M Y, QI D, et al. Salvianolic acid A as a multifunctional agent ameliorates doxorubicin-induced nephropathy in rats[J]. Sci Rep, 2015, 5: 12273.
[5] CHEN Y C, YUAN T Y, ZHANG H F, et al. Salvianolic acid A attenuates vascular remodeling in a pulmonary arterial hypertension rat model[J]. Acta Pharmacol Sin, 2016, 37(6):772-782.
[6] 邱伟文, 郑丽云, 邬至平, 等. 脑心通对氧化低密度脂蛋白诱导血管平滑肌细胞增殖和自噬的影响[J]. 温州医科大学学报, 2016, 46(5): 340-343.
[7] SHADEL G S, HORVATH T L. Mitochondrial ROS signaling in organismal homeostasis[J]. Cell, 2015, 163(3): 560-569.
[8] ROWLANDS D J. Mitochondria dysfunction: A novel therapeutic target in pathological lung remodeling or bystander?[J]. Pharmacol Ther, 2016, 166: 96-105.
[9] LEE J, GIORDANO S, ZHANG J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling[J].Biochem J, 2012, 441(2): 523-540.
[10] GUIDARELLI A, FIORANI M, CERIONI L, et al. Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition[J]. Biofactors,2017, 43(5): 673-684.
[11] MAO K, SHU W, QIU Q, et al. Salvianolic acid A protects retinal pigment epithelium from OX-LDL-induced inflammation in an age-related macular degeneration model[J].Discov Med, 2017, 23(125): 129-147.
[12] PIANTADOSI C A, SULIMAN H B. Mitochondrial dysfunction in lung pathogenesis[J]. Annu Rev Physiol, 2017,79: 495-515.
[13] XIAO B, GOH J Y, XIAO L, et al. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin[J]. J Biol Chem,2017, 292(40): 16697.
[14] WEI X, QI Y, ZHANG X, et al. Cadmium induces mitophagy through ROS-mediated PINK1/Parkin pathway[J]. Toxicology Methods, 2014, 24(7): 504-511.
[15] FENG J, CHEN X, GUAN B, et al. Inhibition of peroxynitrite-induced mitophagy activation attenuates cerebral is chemia-reperfusion injury[J]. Mol Neurobiol (2018). https://doi.org/10.1007/s12035-017-0859-x.
[16] TANG X, CHEN N, LONG X. Ginsenoside Rg1 improves is chemic brain injury by balancing mitochondrial biogenesis and mitophagy[J]. Trop J Pharm Res, 2017, 16(10): 2469-2475.
[17] 郑丽云, 黄慧芬, 邱伟文. 红景天苷对缺氧缺糖诱导的星形胶质细胞线粒体自噬的影响[J]. 温州医科大学学报,2017, 47(10): 744-747.
猜你喜欢
安家(校外教育)(2022年6期)2022-01-03 11:47:06
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:42
建材发展导向(2021年11期)2021-07-28 06:57:22
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
当代水产(2020年10期)2020-03-17 07:02:48
当代水产(2019年8期)2019-10-12 08:57:26
中国药理学通报(2014年2期)2014-05-09 08:22:33
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
中国海洋大学学报(自然科学版)(2014年8期)2014-02-28 12:21:24
