
基于CRISPR-Cas9的功能基因筛选研究进展
2018-05-10 09:31李欢欢黄承浩
生物工程学报 2018年4期
李欢欢,黄承浩
厦门大学生命科学学院 分子疫苗学和分子诊断学国家重点实验室 国家传染病诊断试剂与疫苗工程技术研究中心,福建厦门 361102
随着科学技术的发展,人类基因组计划初步完成,人们对基因的功能也有了一定的认识,但是仍然有更多的问题亟待探索,例如基因与疾病发生发展的关系,基因与肿瘤耐药性的关系等。如何利用人类基因组以及已有的技术手段研究疾病发生发展过程,寻找与疾病和健康相关的功能性基因,从而建立新的疾病诊断预防和治疗方法,发现新的治疗靶点及开发新的药物,是科学家们关注的热点。高通量功能性基因筛选以基因编辑技术为基础,通过在全基因组范围内干扰基因功能来筛选目的表型,是剖析基因功能、探索生物进程和疾病的重要工具。近年来,随着基因编辑技术的发展和成熟,功能性基因筛选也随之变得更加简便高效,在生命科学的研究中发挥了重要作用。本文主要介绍应用CRISPR-Cas9文库进行功能性基因筛选的方法及目前的研究进展。
筛选功能性基因的常用策略主要分为功能获得型 (Gain-of-function)筛选和功能缺失型 (Lossof-function)筛选。功能获得型筛选策略通常利用cDNA文库使基因过表达来筛选功能性基因[1]。由于细胞转录组非常复杂,难以构建覆盖整个细胞基因组的cDNA文库,导致cDNA文库的库容量有限;此外,cDNA的表达并不受细胞内在调控,因此经常会出现基因异常高表达,影响对结果的判断,这些都限制了cDNA文库在功能性基因筛选中的应用。功能缺失型 (Loss-of-function)筛选策略是通过抑制基因表达或基因敲除来筛选功能性基因。RNAi是真核生物细胞内广泛存在的生物进程,通过siRNA介导其同源RNA降解实现抑制靶基因表达的作用[2],因而曾被广泛应用于高通量的功能缺失型基因筛选[3]。与cDNA文库相比,RNAi文库筛选具有高通量、简便、经济、高效的优点,但是其仍然存在难以忽视的问题。RNAi技术是降低基因表达水平来影响表型,并不能完全抑制基因表达,因此其造成的表型变化不够稳定明显,影响对基因功能的判定[4]。此外,细胞内源性的RNAi途径导致RNAi筛选存在着广泛的脱靶效应[5],成为影响RNAi文库筛选的另一个重要问题。近年来CRISPR-Cas9技术大放异彩,可以用于酵母菌[6]、哺乳动物细胞[7]、斑马鱼[8]、水稻[9]、小鼠[10]等各类细胞及生物体的基因编辑,科学家们也开始利用CRISPR-Cas9技术进行功能性基因的筛选。CRISPR-Cas9技术既可通过诱导基因突变进行功能缺失型 (Loss-offunction)筛选,又可通过激活转录进行功能获得型 (Gain-of-function)筛选,不仅可以作用于基因的编码区,还可以靶向基因非编码区[11],与RNAi技术相比具有相对较低的脱靶效率和更广泛的作用位点,因而逐渐成为功能性基因筛选的热点方法。
1 CRISPR-Cas9:精准的基因编辑技术
CRISPR-Cas是广泛存在于细菌和古菌中的获得性免疫系统,能够剪切外源基因,抵御病毒对细菌的入侵[12],最早由Ishino等发现于1987年[13],直到2010年CRISPR的机制和功能才被研究清楚[14-16]。目前被发现的CRISPR系统有3种类型,均由3种元件组成:一簇CRISPR相关基因 (Cas)、非编码RNA以及一列重复序列[17]。Cas基因可以和核酸酶、解旋酶、聚合酶结合,是CRISPR-Cas系统中执行剪切功能的元件[18]。一系列短重复序列被来源于外源基因的间隔序列 (Protospacer)隔开,共同构成了CRISPR RNA(crRNA)阵列。通常间隔序列与前间区序列邻近基序 (PAM)相连[19]。不同物种PAM序列不同,例如目前在哺乳动物细胞中应用广泛的spCas9识别5′-NGG PAM序列,而在细胞基因组中平均8–12 bp就有这样的序列出现[20-21]。基因组中广泛存在的PAM序列为CRISPR-Cas9基因编辑技术应用于全基因组提供了可能。基因座上游是一段几百bp的非编码基因,被命名为前导序列[18]。CRISPR-Cas系统中最常用于基因编辑的是Ⅱ型CRISPR-Cas系统,由Cas9核酸酶、crRNA和反式激活crRNA(tracrRNA)三部分组成[22]。执行基因剪切功能时,首先是crRNA转录为pre-crRNA,同时与crRNA互补的tracrRNA也进行转录并激活Cas9及特异性的RNA核酸酶对pre-crRNA进行加工,成熟的crRNA与tracrRNA及Cas9核酸酶形成复合体,进而在向导RNA的指导下靶向特定位点进行切割[23]。研究者们将密码子优化的Cas9和必要的RNA元件在哺乳动物细胞中进行异源重组表达[24],并将crRNA和tracrRNA融合表达形成一条嵌合单链向导RNA(sgRNA)[25],精简了CRISPR-Cas9系统的结构,极大地提高了CRISPR-Cas9系统应用于基因编辑的可行性,使其更加便捷高效。在CRISPR-Cas9系统中,sgRNA和紧接着sgRNA的PAM是决定基因编辑准确性的关键因素。sgRNA长20 nt,通过与靶序列互补配对引导Cas9准确定位于靶基因,并在PAM上游约3 bp处进行DNA双链剪切[17]。断裂双链 (DSBs)的DNA损伤修复途径主要有以下两种:缺少修复模板时,断裂双链通过非同源末端连接 (Nonhomologous end joining,NHEJ)途径重新连接,这种过程容易产生插入或缺失突变使基因功能丧失,因而被应用于基因敲除;当存在同源修复模板时,断裂双链通过同源直接修复 (Homology-directed repair,HDR)途径将修复模板重组进断裂部位,常被用于基因重组[20]。CRISPR-Cas9技术除了可以进行基因敲除,还可以用于基因的上调或下调表达。CRISPR干扰 (CRISPR interference,CRISPRi)和RNAi功能相似,是在CRISPR技术基础上改造而来的,将Cas9突变使其丧失活性 (dead Cas9,dCas9)无法对双链DNA进行切割,再与转录抑制因子联合作用,即可在sgRNA指导下抑制特定基因的表达[26]。CRISPR激活 (CRISPR activation,CRISPRa)是利用dCas9和转录激活因子协作上调靶基因表达水平,可以被应用于功能获得型筛选[27]。
2 CRISPR-Cas9基因文库
2.1 基因文库设计
目前研究者们已对如何将CRISPR-Cas9系统应用于功能性基因筛选进行了多种尝试并取得了成功 (表1),为后来者提供了大量的经验和指导。CRISPR-Cas9功能性基因筛选平台首先需要有能够满足实验者需求的CRISPR-Cas9文库。有效的sgRNA是决定CRISPR-Cas9功能性基因筛选成功的关键因素,因此首先要设计针对全基因组的sgRNA。设计sgRNA要考虑两点:一是Cas9作用必需的PAM序列,二是降低脱靶效率[17]。研究者们开发了多种用于设计sgRNA的平台。Chuai等[28]总结了当前的36个设计sgRNA的网站,并根据平台设计规则将其分为3类:1)Alignment-based:单纯依据PAM位置设计及评分;2)Hypothesisdriven:依据GC含量、外显子位置等因素对sgRNA效率的作用设计及评分;3)Learningbased:利用考虑影响sgRNA效率的不同特点的训练模式设计及评分。其中第二、三类平台在设计时考虑了不同序列和染色质特点,相比第一类平台更加全面高效,使用者可根据不同要求选择适合的平台。设计好的sgRNA合成后需要连接至合适的慢病毒载体上,Cas9核酸酶既可以和sgRNA构建在同一个质粒上[29],也可以构建在两个质粒上[30]。将构建好的sgRNA文库包装成慢病毒,并以低MOI(通常为0.3–0.5)感染细胞构建稳定细胞株[29],避免同一细胞多个基因同时被敲除对结果造成影响。
2.2 常用筛选方式
CRISPR-Cas9文库高通量筛选常用模式可以分为阳性筛选 (Positive selection screen)和阴性筛选 (Negative selection screen)[30]。阳性筛选是对已成功整合sgRNA的细胞文库施加一定的筛选压力,仅使少数目的表型的细胞能够存活,达到富集关键基因的目的。阴性筛选与之相反,存活的细胞并不是目的表型细胞,因此需要比较不同时间点sgRNA的丰度找出差异sgRNA来确定关键基因。两种筛选方式通常都需要与高通量测序技术结合,通过高通量测序获得样品中细胞的sgRNA序列信息,再通过生物信息学分析排除假阳性结果继而确定可能的候选基因 (图 1)。CRISPR系统后续分析同样是影响实验结果的重要问题,Chuai等[28]同样整理了7种用于后续结果分析的平台。例如MAGeCK是Li等[31]开发的用来分析CRISPR高通量筛选结果的算法,之后Li等又将其拓展为MAGeCK-VISPR算法[32],为CRISPR高通量筛选提供了综合质控、分析及可视化的数据分析方法。
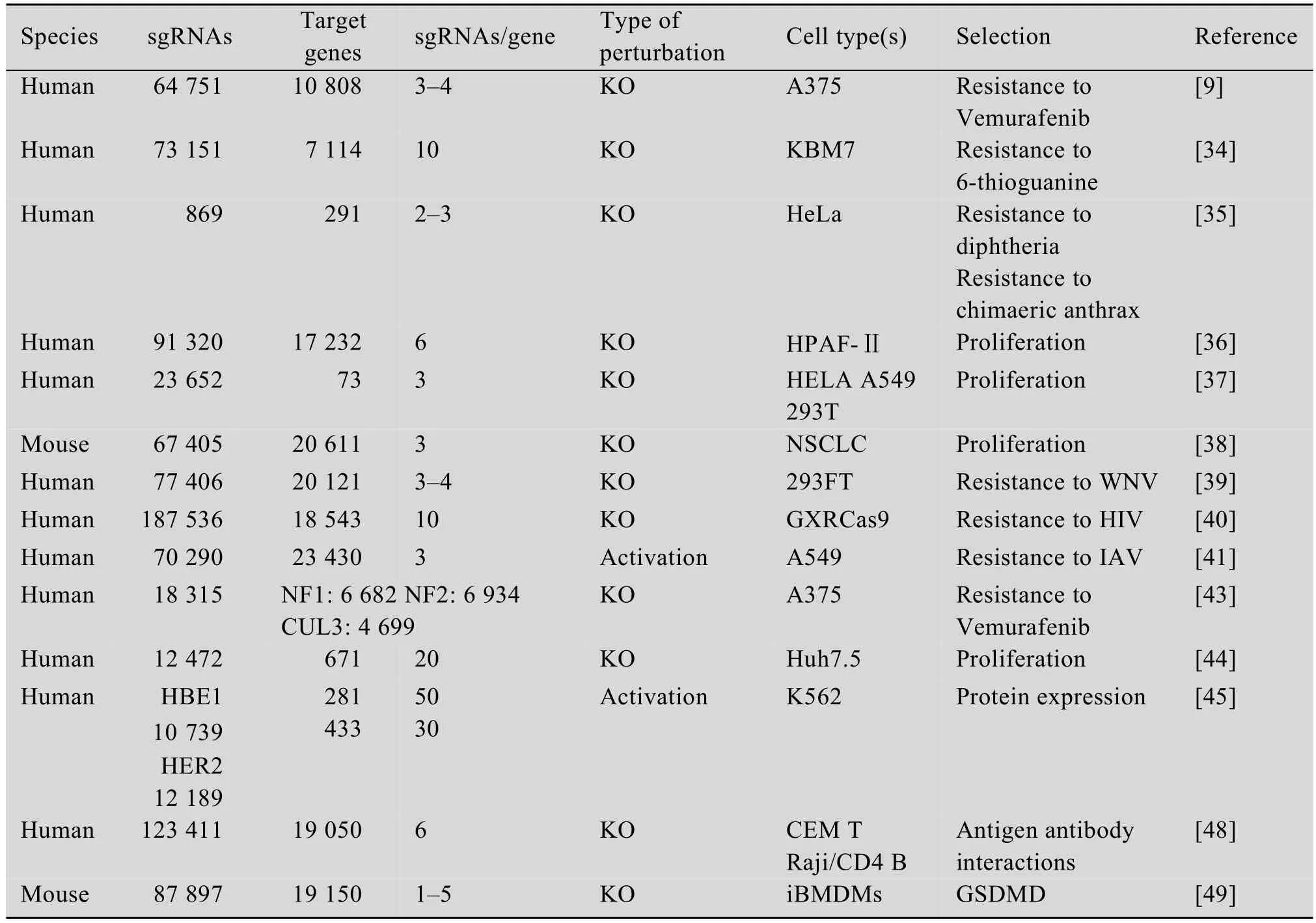
表1 CRISPR-Cas9系统在功能基因筛选中的应用Table 1 Functional genetic screening using CRISPR-Cas9 system
3 CRISPR-Cas9文库应用于功能性基因筛选的研究进展
3.1 药物作用基因筛选
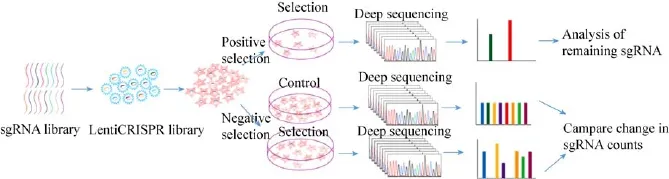
图1 CRISPR-Cas9系统功能基因筛选流程Fig.1 Flowchart of functional genetic screening using CRISPR-Cas9 system.
CRISPR-Cas9文库早期应用于药物作用基因筛选的尝试较多。Vemurafenib是突变的蛋白激酶BRAF的抑制剂,被批准用于晚期黑色素瘤的治疗,对存在V600E BRAF突变的黑色素瘤有治疗效果。Shalem等[29]设计了靶向全基因组18080个基因,包含64751条sgRNA的CRISPR-Cas9基因敲除文库,并命名为GeCKO(Genome-scale CRISPR-Cas9 knockout)文库。Shalem等利用慢病毒载体将GeCKO文库转染进有V600E BRAF突变的黑色素瘤细胞A375中,加入Vemurafenib抑制A375细胞生长,从而富集少数具有Vemurafenib药物抗性的细胞。通过对这部分细胞的sgRNA序列进行分析,Shalem等筛选出了nf1、med12、nf2、cul3、tada2b和tada1等敲除后能够使细胞对Vemurafenib产生抗性的基因,其中nf2、cul3、tada2b和tada1之前尚未被报道过与Vemurafenib耐药性有关。这些候选基因提出了新的Vemurafenib肿瘤耐药机制假说。研究者还将筛选结果与之前使用RNAi[33]筛选Vemurafenib耐药基因的结果进行比较评估,两者结果高度一致,证明了CRISPR-Cas9用于功能性基因筛选的可行性。
同期,Wang及其团队也采用了类似的筛选方式研究了DNA错配修复的相关基因[34]。研究者使用能够引起细胞损伤的核苷类似物6-硫代鸟嘌呤作为筛选抗性,MMR通路正常的细胞无法修复6-硫代鸟嘌呤引起的损伤而导致细胞滞留在G2-M期无法进行正常的分裂,而MMR缺陷细胞则不能识别这种损伤从而继续进行分裂。Wang等设计合成了靶向7114个基因的sgRNA文库,并且每个基因至少设计了10条sgRNA以确保文库的knockout效率。与Shalem的方法略有差别的是,Wang等并未采用单质粒的慢病毒载体系统,而是先构建稳定表达Cas9的KBM7细胞,再转染了整个sgRNA文库。与单质粒系统相比,双质粒系统可以通过挑选高效的Cas9表达细胞株来提高文库的knockout效率。完成建库工作后研究者利用6-硫代鸟嘌呤的细胞致死作用,杀死MMR通路正常细胞,富集MMR通路突变细胞。对存活细胞的sgRNA测序分析,研究者发现被富集细胞的sgRNA主要靶向MMR通路的4个关键基因msh2、msh6、mlh1和pms2。
sgRNA文库的构建是实现功能性基因筛选的关键。覆盖全基因的文库虽然更加全面,但是由于库容量庞大,在实际操作过程中需要大量的细胞,工作量及操作难度均较大。在研究炭疽毒素与白喉毒素毒性作用相关基因的工作中,Zhou等[35]通过对已掌握知识进行整理,挑选出了可能与研究内容相关的291基因,并设计了靶向这291个基因的包含869条sgRNA的慢病毒聚焦型人源文库。该课题组使用炭疽毒素和白喉毒素分别进行阳性筛选,经过三轮毒素处理,待对照组细胞全部死亡后,对实验组存活细胞进行测序分析。除了已知的炭疽毒素受体Antxr1,研究者还成功鉴定plxna1、pecr、fzd10和CD81等与炭疽毒素作用机制相关的基因。已知白喉毒素相关基因hbegf及新的与白喉毒素相关基因rab2a也被富集。这种聚焦型的文库缩小了筛选范围,降低了操作难度和实验成本,尤其适用于研究者对筛选功能基因已有一定了解的情况。根据实际情况选择适合的文库容量能够更有效地进行文库筛选工作,提高实验成功率。
3.2 肿瘤功能基因筛选
肿瘤是机体在遗传及环境等多重因素的影响下,致癌基因和抑癌基因发生突变而导致的细胞生长不受调控的疾病,严重威胁着人类的生命健康,引起了科学家的高度重视。运用高通量筛选技术鉴别参与肿瘤发生发展及转移的关键基因是研发抗肿瘤新药的重要手段。CRISPR文库也被应用于肿瘤治疗靶点和新型抗肿瘤药物筛选,取得了良好的成果。
胰腺癌生长快、转移率高且预后差,被称为“癌中之王”,其中胰腺导管腺癌 (PDA)是胰腺癌的一个主要的类型。Steinhart等[36]选取了一株RNF43突变的胰腺导管腺癌细胞 (PDAC)HPAF-Ⅱ来筛选胰腺癌新的治疗靶标。Steinhart及其团队中将靶向全基因组17232个基因的TKO gRNA文库转导进HPAF-Ⅱ细胞,通过对不同时间点细胞的sgRNA的丰度变化进行分析从而鉴别影响细胞增殖的基因。Steinhart等鉴别出了2174个影响HPAF-Ⅱ细胞增殖的基因,其中参与Wnt信号通路的FZD5,对PDAC的增殖具有重要的意义。随后研究者还发现特异性结合FZD5和FZD8的抗体能够抑制RNF43突变PDAC细胞生长,在肿瘤异种移植模型中也有类似作用,并且对RNF43突变的结直肠癌也有抑制效果。因此Steinhart等提出FZD5可以作为胰腺癌的潜在治疗靶点,基于CRISPR的高通量功能性基因筛选是筛选细胞表面特异性治疗靶标的有效手段。
一些抗肿瘤药物仅对发生特异性基因突变的细胞起作用,其作用机制是当肿瘤细胞中某个基因发生突变时抑制另一基因表达,因而能够特异性地杀伤突变率高的肿瘤细胞而不影响正常细胞,这一作用方式被称为合成致死性。Shen等[37]从这一思路出发,设计了双重gRNA文库筛选合成致死相互作用网络。与一般的sgRNA文库不同,双重gRNA文库中每个载体包含两条gRNA,一条靶向肿瘤细胞中高突变的肿瘤抑制基因,另一条靶向一种能够被肿瘤治疗药物抑制的基因。Shen等设计了靶向73对癌症基因,共计141912种基因组合的双重gRNA文库,并在人宫颈癌细胞、肺癌细胞和人胚肾细胞中进行了筛选,通过检测不同时间点gRNA丰度变化,进一步分析筛选出120种合成致死相互作用,为癌症新药的开发提供了新的靶点。
CRISPR-Cas9功能性筛选技术也是研究肿瘤生长与转移的重要工具之一。Chen等[38]用包含67405条sgRNA的基因文库诱变处理一种非转移的鼠非小细胞肺癌细胞株,并将其移植于免疫缺陷鼠身上,发现肿瘤很快就出现了转移。Chen等将肺转移细胞与原发部位细胞进行测序比较sgRNA丰度变化,筛选出nf2、pten、cdkn2a、trim72、fga、miR-345及miR-152七个与肿瘤转移相关的基因。Chen等的工作证明了CRISPR-Cas9全基因组筛选不仅可以在各种细胞中开展,并且在体内也同样适用。Chen及其团队所提供的完整的体内筛选方案在之后也被更广泛地应用于各种疾病的体内研究工作。与体外筛选相比,动物体内筛选模型具有更复杂的微环境,比体外更接近真实的肿瘤微环境,从而有可能筛选到依赖于肿瘤微环境的肿瘤抑制基因。
3.3 病毒感染基因筛选
许多疾病都与病毒感染相关,筛选出病毒受体或病毒在宿主细胞中复制关键基因有助于开发新的传染病预防及治疗策略。西尼罗病毒 (West Nile virus,WNV)是一种脑炎病毒,能够引起急性神经感染并导致大量神经元细胞死亡。为了研究WNV导致细胞死亡的机制,Ma及其团队[39]构建了靶向人类基因组20121个基因的sgRNA文库,并将每条sgRNA的靶点都设计在转录起始位点附近以确保基因能够被完全敲除。Ma等将构建好的文库转导进293FT细胞中,并感染WNV病毒株进行了第一轮筛选。Ma等对第一轮筛选结果进行分析,发现有部分丰度较高的sgRNA其靶向基因的其他sgRNA并未被富集,这些结果可能是由假阳性造成的。因此Ma及其团队又针对初次实验结果富集到的sgRNA设计了亚基因敲除文库进行重复实验,最终筛选出7个与WNV诱导细胞死亡相关的关键基因:emc2、emc3、sel1l、derl2、ubeg2、ube2j1和hrd1。研究者证实了敲除这些基因能够有效保护细胞不被WNV杀死,这7个基因可以作为新的治疗靶点以降低WNV导致的机体损伤。
HIV引发的艾滋病 (AIDS)严重威胁着人类生命健康,阐明HIV突破宿主细胞防御系统的分子机制,开发HIV治疗的新靶点,进而提出新的HIV防治策略,具有非常重大的理论意义和实际应用价值。Park等[40]为了阐明HIV在T细胞上的受体,设计了适用于CRISPR筛选的CD4+T细胞(GXRCas9细胞)筛选模型,并用包含187536条sgRNA(靶向18543个基因)的慢病毒文库感染该细胞,获得了超过18万种缺失不同受体的T细胞库。之后研究人员用HIV病毒株JR-CSF感染这些缺失不同受体的T细胞,随后通过流式分选GFP阴性和阳性的T细胞 (被病毒感染的细胞能够检测到GFP的表达),并对GFP阴性细胞群与未感染HIV病毒细胞测序,分析两群细胞sgRNA的丰度差异,最终发现了5个sgRNA丰度变化最大的基因。其中CD4和CCR5是HIV感染T细胞的受体,酪蛋白磺基转移酶2(Tyrosylprotein sulfotransferase 2,TPST2)和溶质家族35成员B2(Solute carrier family 35 member B2,SLC35B2)对CCR5进行修饰以便于与HIV结合。另一种基因编码白细胞粘附因子 (Activated leukocyte cell adhesion molecule,ALCAM),与HIV在细胞间的传播相关。筛选出的5个基因被敲除后均不影响T细胞的存活,但能够使T细胞抵抗HIV的感染,因此可以作为HIV治疗的潜在靶标,为预防和治疗艾滋病提供新的理念和途径。
目前功能缺失型 (Loss-of-function)筛选已被广泛应用于寻找病毒感染、复制和传播所需基因,但过表达基因影响病毒感染的研究还比较欠缺。基于此Heaton等[41]应用CRISPR协同激活调节(CRISPR synergistic activation mediator,CRISPR SAM)技术筛选过表达后抑制甲型流感病毒 (IVA)感染的基因。研究者首先将dCas9-VP64融合蛋白和MS2-p65-HSF1转录因子转导进A549-CR细胞中,并将靶向全基因组23430个基因转录起始位点上游200 bp处的sgRNA文库导入转录激活效率最高的细胞株,促进基因的过表达。之后研究者用IVA感染细胞,通过流式分选出未被病毒感染的细胞进行测序分析,发现靶向b4galnt2基因的sgRNA丰度最高。经过多重验证,Heaton等证明了b4galnt2过表达能够限制多种亚型的甲型流感病毒感染,未来可能被应用于防治人流感及禽流感。
为了研究影响溶瘤病毒感染及杀伤肿瘤细胞的关键基因,我们同样采用基于GeCKO v2全基因组敲除文库的功能缺失型 (Loss-of-function)筛选方法。我们尝试了两种筛选方案:第一种是阵列筛选 (Arrayed screen),即将已成功转导GeCKO v2文库的病毒抵抗型细胞分入96孔板,通过免疫斑点法检测病毒复制情况,挑选病毒复制提高的孔进行下一步验证;第二种方法是合并筛选 (Pooled screen),即将成功转导GeCKO v2文库的细胞感染病毒,利用高通量测序分析病毒感染后细胞sgRNA丰度变化情况。与合并筛选相比,阵列筛选每孔细胞数较少,因此在培养过程中易出现sgRNA丢失的情况,同时实验结果还与用于检测病毒复制抗体的亲和性有关,容易出现假阳性,降低了实验的准确性,且该方法工作量较大,操作难度高。合并筛选是较为便捷准确的筛选方式,需要注意的是筛选所用细胞的选择,可依据正向筛选和负向筛选分别选择病毒敏感型细胞和病毒抵抗型细胞。
3.4 非编码基因功能研究
基因组中大多数DNA并不编码蛋白质,但却与基因表达调控密切相关,研究表明超过90%遗传变异相关疾病发生在基因非编码区域[42]。阐明这些非编码基因如何参与基因表达调控有助于人们更好地研究基因异常导致的疾病。在之前的工作中,Shalem等[29]利用CRISPR-Cas9文库发现了nf1、nf2、clu3等功能缺失后使黑色素瘤细胞对Vemurafenib产生抗性的基因。在此基础上,Sanjana等[43]设计了3个分别靶向cul3、nf1和nf2基因5′端和3′端紧邻的100 kb范围内基因区域的sgRNA文库,来研究非编码区对相关基因的调控机制。Sanjana等将sgRNA文库转导进BRAF突变的A375细胞中,将Vemurafenib处理后的存活细胞测序分析。研究者对富集sgRNA最多的cul3基因进行了重点分析,发现在cul3基因周围有24个基因位点突变能够导致cul3表达降低。Sanjana及其团队的工作证明了CRISPR-Cas9文库不仅可以用于筛选功能性基因,还可以用来研究非编码基因功能。
对于非编码长链RNA(lncRNA),插入或缺失突变导致的变化可能并不足以产生明显的表型变化。针对这一情况,Zhu等[44]设计了配对向导RNA(Paired-guide RNA,pgRNA),对人肝癌细胞系Huh7.5OC中的近700个癌症或其他疾病相关长链非编码RNA的基因进行了功能筛选。pgRNA是一对靶向不同位点的sgRNA,能够在同一基因的不同位点造成双链断裂,因此有可能产生大片段的缺失,造成可观察的表型变化。为了验证pgRNA的有效性,研究人员设计了靶向cspg4基因座的pgRNA,使用双U6启动子分别启动两个gRNA的表达,结果表明所有的6对pgRNA均能够对基因正确切割。基于此,Zhu等针对Huh7.5OC中的近700个癌症或其他疾病相关长链非编码RNA设计了gRNA,剔除其中特异性和效率较低的gRNA及靶向启动子和外显子的gRNA,获得了可用于筛选的基因文库。之后研究者将文库转导进稳定表达Cas9的Huh7.5OC,持续培养30 d后对CRISPR筛选前和筛选后的细胞测序分析来鉴定能够影响细胞增殖和生存的lncRNA。经过多重的验证,Zhu等鉴定出了51个与肿瘤细胞增殖相关的非编码RNA,证实了利用pgRNA研究lncRNA的方法具有较高的精确性和特异性。同时,研究人员在Hela细胞中的重复实验结果也证明了这一点。但是这一筛选策略仍存在一些问题。首先删除lncRNA可能影响邻近功能元件如增强子和microRNA的功能,因此需要在设计pgRNA时尽可能地排除靶向增强子和microRNA区域的gRNA。由于pgRNA间具有相同的U6启动子和3′支架序列,不同的pgRNA间有可能 (约7.5%)会发生重组导致错误的配对,使用不同的U6启动子和3′支架序列可以降低重组率。此外,这种筛选策略并不能揭示lncRNA的调节机制,还需要后续的工作进行探究。
Klann及其团队[45]介绍了一种以CRISPRCas9技术为基础的表观遗传调节元件筛选方法(CERES)。识别调节元件所有可能的转录因子结合位点需要针对该调节元件区域设计高密度的sgRNA。然而在许多基因区域PAM序列可能并不在转录因子结合位点,导致sgRNA不能充分覆盖所有的转录因子结合位点。因此Klann等选用了转录抑制的dCas9KRAB酶和转录激活的dCas9p300酶。dCas9KRAB 是将 KRAB(Krüppelassociated box)区域与dCas9融合表达,招募使组蛋白H3K9发生甲基化的蛋白进而形成异染色质导致靶序列上的基因抑制[46]。E1A相关蛋白p300组蛋白乙酰转移酶核心区与dCas9融合后能够结合到靶DNA增强子或启动子上,促进组蛋白H3K27发生乙酰化,从而促使基因激活[47]。这两种方法即使在转录因子结合位点没有PAM序列的情况下仍然能够对调节元件活性进行调节。研究人员根据人白血病细胞系K562 DNase-seq数据设计了针对b-globin基因座周围4.5 Mb区域的10739条sgRNA。这些sgRNA靶向该区域281个DNaseⅠ超敏感位点 (DNaseⅠhypersensitive sites,DHSs)。研究者将构建好的sgRNA导入改造过的细胞 (b-globin的转录与荧光蛋白相关联)中,并分别将荧光表达最高和最低的10%细胞分选出来分析sgRNA的相对丰度变化。Klann及其团队的工作为高通量注释调节元件的作用提高了效率。
3.5 其他工作
除了应用于肿瘤、病毒、非编码RNA的相关功能基因研究外,CRISPR-Cas9高通量筛选在抗体靶标筛选、细胞信号通路研究等生物医学的其他领域中也有着广泛的应用。
单克隆抗体特异性靶标抗原及其表位的识别是抗体研究中的重要工作。传统的抗原特异性鉴别方式是Western blotting(蛋白印迹)和Immunoprecipitation(免疫沉淀),但是当抗体不能被Western blotting和Immunoprecipitation检测到时,就需要使用基于基因的技术来鉴定抗体靶标。Zotova等[48]以MT2细胞 (人T细胞嗜淋巴病毒Ⅰ型HTLV-1慢性感染T细胞)作为免疫原免疫小鼠获得了一株HTLV-1生物膜特异性单克隆抗体BF4。BF4能够和未感染的淋巴细胞、中性粒细胞及HTLV-1感染细胞表面的病毒生物膜结合。Zotova等的研究思路是在BF4阳性细胞上转导CRISPR knockout文库,分选出阴性细胞,即可能是BF4抗原被敲除的细胞。基于这样的思路研究者在CEM T和Raji/CD4 B细胞上转导GeCKO文库,并将不与BF4结合的细胞分选出来。经过两轮的重复分选,阴性细胞比例达到了99%以上。研究人员对这部分细胞进行测序分析,发现约80%的sgRNA靶向CD82,经过验证,证实了BF4确实是CD82特异性抗体。在筛选抗体靶标的研究中还有一些需要注意的地方,首先这种筛选方式不适合筛选抗体抗原是细胞生存所需的基因,因为这部分基因被敲除后会导致细胞无法存活而丢失,无法在后续的分选工作中被鉴定出。Cas9文库的代表性和多样性也是需要关注的问题,不同的细胞和慢病毒载体都会对此造成影响。
Shi及其团队[49]在Nature上发表了关于细胞焦亡机制的相关研究。细胞焦亡是机体在感知病原微生物浸染后启动的免疫防御反应,炎症激活的caspase-1和识别细菌脂多糖的caspase-4、caspase-5和caspase-11都能够引起细胞焦亡,但是其机制仍然未知。研究人员首先建立了脂多糖(LPS)电转方法,使LPS能够诱导90%以上的细胞焦亡。接着在能够对脂多糖刺激正常反应的Tlr4–/–骨髓源永生化巨噬细胞 (iBMDMs)转导CRISPR knockout基因文库,将脂多糖刺激细胞焦亡后存活的细胞测序分析。分析结果显示,靶向gasdermin D(GSDMD)基因的5条sgRNA中,有4条在富集到的前30中,其中有2条在前10。后续的结果进一步证明了GSDMD的N端能够诱导细胞焦亡。在此研究中,研究人员利用CRISPR基因文库进行了全基因组范围的遗传筛选,成功地筛选到了敲除后能够抑制细胞焦亡的基因GSDMD,并阐明了GSDMD作为炎症caspase底物蛋白,在被切割后能够引发细胞焦亡的分子机制。
4 结语
CRISPR-Cas9技术作为近年来热门的基因编辑技术,凭借其精准、简便、高效的特点受到众多研究者的关注,短短几年间已成为基因编辑领域中最热门的技术,被广泛应用于生物、医学等领域。基于CRISPR-Cas9的高通量功能性筛选技术也逐渐取代了RNAi和cDNA文库,为功能基因的研究提供了高通量高效率的筛选工具。尽管与传统的筛选方法相比,CRISPR-Cas9更加高效可靠且特异性更高,但其仍然存在一些问题,其中最令人关注的就是脱靶效率。CRISPR-Cas9系统依据20 nt的sgRNA定位靶基因,因此有可能由于错配而出现脱靶问题。除了设计特异性更高的sgRNA外,研究者们提供了其他的降低脱靶效率的策略。例如可以使用只切开DNA双链一侧的Cas9n切口酶[50],需要一对sgRNA才能完成DNA双链剪切,因此可以降低脱靶效率。Vidigal等[51]则是将20 nt的sgRNA缩短为17 nt,剪切效率不变而脱靶效率降低。另一种策略是将dCas9与FokⅠ核酸酶融合表达来提高筛选特异性[52]。这些方法虽然能够降低脱靶效率,但是还需要进一步改进才能应用于高通量的功能性基因筛选。
基于CRISPR-Cas9功能性基因筛选技术已经在细胞生存必需基因鉴定、药物或毒素抗性基因筛选、潜在治疗靶标筛选、肿瘤转移等相关基因筛选方面取得了重要进展,为开发有效的靶点特异性治疗药物奠定了基础,未来也必然会推动整个生命科学的快速发展。
[1]YangXP,Boehm JS,YangXP,etal.A public genome-scale lentiviral expression library of human ORFs.Nat Meth,2011,8(8):659–661.
[2]Rana TM.Illuminating the silence:understanding the structure and function of small RNAs.Nat Rev Mol Cell Biol,2007,8(1):23–36.
[3]Mohr SE,Smith JA,Shamu CE,et al.RNAi screening comes of age:improved techniques and complementary approaches.Nat Rev Mol Cell Biol,2014,15(9):591–600.
[4]Sachse C,Krausz E,Krönke A,et al.High-throughput RNA interference strategies for target discovery and validation by using synthetic short interfering RNAs:functional genomics investigations of biological pathways.Methods Enzymol,2005,392:242–277.
[5]Qiu SB,Adema CM,Lane T.A computational study of off-target effects of RNA interference.Nucleic Acids Res,2005,33(6):1834–1847.
[6]Gratz SJ, Wildonger J, Harrison MM, et al.CRISPR/Cas9-mediated genome engineering and the promise of designer flies on demand.Fly(Austin),2013,7(4):249–255.
[7]Jinek M,East A,Cheng A,et al.RNA-programmed genome editing in human cells.eLife,2013,2:e00471.
[8]Hruscha A,Krawitz P,Rechenberg A,et al.Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish.Development,2013,140(24):4982–4987.
[9]Shan QW,Wang YP,Li J,et al.Targeted genome modification of crop plants using a CRISPR-Cas system.Nat Biotechnol,2013,31(8):686–688.
[10]Fujii W,Onuma A,Sugiura K,et al.Efficient generation of genome-modified mice via offset-nicking by CRISPR/Cas system.Biochem Biophys Res Commun,2014,445(4):791–794.
[11]Shalem O,Sanjana NE,Zhang F.High-throughput functional genomics using CRISPR-Cas9.Nat Rev Genet,2015,16(5):299–311.
[12]Wei CX,Liu JY,Yu ZS,et al.TALEN or Cas9-rapid,efficient and specific choices for genome modifications.J Genet Genomics,2013,40(6):281–289.
[13]Ishino Y,Shinagawa H,Makino K,et al.Nucleotide sequence of the iap gene,responsible for alkaline phosphatase isozyme conversion inEscherichia coli,and identification of the gene product.J Bacteriol,1987,169(12):5429–5433.
[14]Horvath P,Romero DA,Coûté-Monvoisin AC,et al.Diversity,activity,and evolution of CRISPR loci inStreptococcus thermophilus.J Bacteriol,2008,190(4):1401–1412.
[15]Horvath P,Coûté-Monvoisin AC,Romero DA,et al.Comparative analysis of CRISPR loci in lactic acid bacteria genomes.Int J Food Microbiol,2009,131(1):62–70.
[16]Garneau JE,DupuisMÈ,Villion M,etal.The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA.Nature,2010,468(7320):67–71.
[17]Ran FA,Hsu PD,Wright J,et al.Genome engineering using the CRISPR-Cas9 system.Nat Protoc,2013,8(11):2281–2308.
[18]Karginov FV,Hannon GJ.The CRISPR system:small RNA-guided defense in bacteria and archaea.Mol Cell,2010,37(1):7–19.
[19]Grissa I,Vergnaud G,Pourcel C.The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats.BMC Bioinform,2007,8:172.
[20]Cong L,Ran FA,Cox D,et al.Multiplex genome engineering using CRISPR/Cas systems.Science,2013,339(6121):819–823.
[21]Hsu PD,Scott DA,Weinstein JA,et al.DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol,2013,31(9):827–832.
[22]Gasiunas G,Barrangou R,Horvath P,et al.Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria.Proc Natl Acad Sci USA,2012,109(39):E2579–E2586.
[23]Jinek M,Jiang FG,Taylor DW,et al.Structures of Cas9 endonucleasesrevealRNA-mediated conformational activation.Science,2014,343(6176):1247997.
[24]Mali P,Yang L,Esvelt KM,et al.RNA-guided human genome engineering via Cas9.Science,2013,339(6121):823–826.
[25]Jinek M,Chylinski K,Fonfara I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.Science,2012,337(6096):816–821.
[26]Gilbert LA,Larson MH,Morsut L,et al.CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes.Cell,2013,154(2):442–451.
[27]Perez-PineraP,Kocak DD,Vockley CM,etal.RNA-guided gene activation by CRISPR-Cas9-based transcription factors.NatMethods,2013,10(10):973–976.
[28]Chuai GH,Wang QL,Liu Q.In silicomeetsin vivo:towards computational CRISPR-based sgRNA design.Trends Biotechnol,2017,35(1):12–21.
[29]Shalem O, Sanjana NE, Hartenian E, et al.Genome-scale CRISPR-Cas9knockoutscreeningin human cells.Science,2014,343(6166):84–87.
[30]Xu H,Xiao TF,Chen CH,et al.Sequence determinants of improved CRISPR sgRNA design.Genome Res,2015,25(8):1147–1157.
[31]Li W,Xu H,Xiao TF,et al.MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens.Genome Biol,2014,15:554.
[32]Li W,Köster J,Xu H,et al.Quality control,modeling,and visualization of CRISPR screens with MAGeCK-VISPR.Genome Biol,2015,16:281.
[33]Whittaker SR,Theurillat JP,van Allen E,et al.A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition.Cancer Discov,2013,3(3):350–362.
[34]Wang T,Wei JJ,Sabatini DM,et al.Genetic screens in human cells using the CRISPR-Cas9 system.Science,2014,343(6166):80–84.
[35]Zhou YX,Zhu SY,Cai CZ,et al.High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells.Nature,2014,509(7501):487–491.
[36]Steinhart Z,Pavlovic Z,Chandrashekhar M,et al.Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuitas a druggable vulnerability ofRNF43-mutant pancreatic tumors.Nat Med,2017,23(1):60–68.
[37]Shen JP,Zhao DX,Sasik R,et al.Combinatorial CRISPR-Cas9 screens forde novomapping of genetic interactions.Nat Methods,2017,14(6):573–576.
[38]Chen SD,Sanjana NE,Zheng KJ,et al.Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis.Cell,2015,160(6):1246–1260.
[39]Ma HM,Dang Y,Wu YG,et al.A CRISPR-based screen identifies genes essential for west-nile-virus-induced cell death.Cell Rep,2015,12(4):673–683.
[40]Park RJ,Wang T,Koundakjian D,et al.A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors.Nat Genet,2017,49(2):193–203.
[41]Heaton BE,Kennedy EM,Dumm RE,et al.A CRISPR activation screen identifies a pan-avian influenza virus inhibitory host factor. Cell Rep, 2017, 20(7):1503–1512.
[42]Thurman RE,Rynes E,Humbert R,et al.The accessible chromatin landscape of the human genome.Nature,2012,489(7414):75–82.
[43]Sanjana NE,Wright J,Zheng KJ,et al.High-resolution interrogation of functional elements in the noncoding genome.Science,2016,353(6307):1545–1549.
[44]Zhu SY,Li W,Liu JZ,et al.Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library.Nat Biotechnol,2016,34(12):1279–1286.
[45]Klann TS,Black JB,Chellappan M,et al.CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome.Nat Biotechnol,2017,35(6):561–568.
[46]Thakore PI,D’Ippolito AM,Song LY,et al.Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements.Nat Methods,2015,12(12):1143–1149.
[47]Hilton IB,D’Ippolito AM,Vockley CM,etal.Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers.Nat Biotechnol,2015,33(5):510–517.
[48]Zotova A,Zotov I,Filatov A,et al.Determining antigen specificity of a monoclonal antibody using genomescale CRISPR-Cas9 knockoutlibrary.JImmunol Methods,2016,439:8–14.
[49]Shi JJ,Zhao Y,Wang K,et al.Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death.Nature,2015,526(7575):660–665.
[50]Ran FA,Hsu PD,Lin CY,et al.Double nicking by RNA-guidedCRISPR Cas9forenhancedgenome editing specificity.Cell,2013,154(6):1380–1389.
[51]Vidigal JA,Ventura A.Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries.Nat Commun,2015,6:8083.
[52]Tsai SQ,Wyvekens N,Khayter C,et al.Dimeric CRISPR RNA-guidedFokⅠnucleases for highly specific genome editing.Nat Biotechnol,2014,32(6):569–576.
猜你喜欢
河池学院学报(2021年1期)2021-07-10
猪业科学(2021年3期)2021-05-21
透析与人工器官(2020年1期)2020-11-16
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
铁道通信信号(2019年8期)2019-10-10
英语文摘(2019年2期)2019-03-30
中华手工(2018年6期)2018-07-17
猪业科学(2018年4期)2018-05-19
中国发展观察(2017年8期)2017-04-26
