
基于溶剂剥离石墨烯修饰电极高灵敏测定阿昔洛韦
2018-05-07 00:40:59梅素容
分析科学学报 2018年1期
罗 静, 利 国,周 艳, 梅素容
(1.华中科技大学同济医学院公共卫生学院环境医学研究所,教育部环境与健康重点实验室,湖北武汉 430030;2.湖北出入境检验检疫局,湖北武汉 430050;3.湖北省疾病预防控制中心,湖北武汉 430079)
2004年,英国曼彻斯特大学的物理学教授海姆和诺沃肖洛夫等首次利用胶带剥离定向石墨的方法成功地分离出稳定的石墨烯[1],近年来石墨烯在电化学检测领域也得到了广泛的应用[2-6]。在电化学检测中所用的石墨烯绝大多数都采用化学氧化剥离的方法来制备,存在操作繁琐(需多步不同温度的化学氧化)、环境不友好(使用大量的氧化剂和还原剂)等不足,而且化学氧化过程极有可能破坏石墨本身的sp2杂化结构,进而影响石墨烯的性能。随着石墨烯制备方法的深入研究,出现了溶剂剥离制备高质量石墨烯的方法[7]。以石墨为原料,选择合适的剥离溶剂,在超声辅助下利用剥离溶剂的插层作用可制备出高质量的石墨烯纳米片,此方法简单易控,所需设备、试剂简单,对环境危害小。与化学氧化剥离法相比,溶剂剥离法制备出的石墨烯sp2杂化结构保持完好,片层小,具有更大的比表面积以及更为优良的导电性,因此,溶剂剥离法是一种廉价、高效、简便的石墨烯制备方法。
阿昔洛韦是一种广泛使用的抗菌药物,目前其检测方法主要有高效液相色谱[8]、化学发光[9]等方法。目前,已有碳纳米管修饰玻碳电极[10]、2-巯基苯并噻唑自组装膜修饰金电极[11]、纳米铜修饰碳糊电极[12]以及聚乙烯吡咯烷酮修饰碳糊电极[13]被报道用于阿昔洛韦的电化学测定。本文以N,N-二甲基吡咯烷酮(NMP)为溶剂,通过超声剥离制备出石墨烯纳米片,并通过挥发溶剂的方法制备出NMP剥离石墨烯薄膜修饰电极;研究了阿昔洛韦在此电极上的电化学行为,发现NMP剥离石墨烯纳米片对阿昔洛韦的电化学氧化有显著的增敏效应,极大地提高了阿昔洛韦的氧化峰电流;研究了pH值、NMP剥离石墨烯用量、富集时间等参数对阿昔洛韦氧化信号的影响,建立了一种阿昔洛韦高灵敏的电化学检测新方法,发展的方法比目前报道的电化学方法[14 - 16]检出限更低。
1 实验部分
1.1 仪器与试剂
电化学实验在CHI 830D电化学工作站上进行(上海辰华仪器有限公司),采用三电极系统:工作电极为玻碳电极或修饰电极,参比电极为饱和甘汞电极,对电极为铂丝。透射电镜和扫描电镜图谱分别在Tecnai G220 和Quanta 200 电子显微镜上获得(荷兰,FEI公司)。
所用试剂均为分析纯,未经纯化而直接使用。阿昔洛韦(中国药品生物制品检定所)溶解在超纯水中配制成1.0 mmol/L的标准溶液,并在4 ℃保存。光谱纯石墨、NMP、乙酸和乙酸钠,均购自国药集团。
1.2 实验方法
1.2.1石墨烯薄膜修饰电极的制备将1.5 g光谱纯石墨加入到300.0 mL NMP中,在KQ-100B型超声波清洗器中(功率100 W)超声剥离12 h,5 000 r/min离心5 min后,收集上层悬浮液作为电极修饰液。玻碳电极(GCE)修饰前先用50 nm的Al2O3粉抛光,清洗干净后,在其表面精确滴涂5.0 μL石墨烯悬浮液,红外灯下烘干,即制得NMP剥离石墨烯修饰电极。
1.2.2电分析方法以0.1 mol/L乙酸缓冲溶液(pH=5.0)作为阿昔洛韦的测定介质,富集3 min后,记录从0.5 V到1.1 V的微分脉冲伏安曲线,测量0.94 V处的峰电流作为阿昔洛韦的响应信号。微分脉冲振幅为50 mV,脉冲宽度为40 ms,扫描速度为40 mV/s。
2 结果与讨论
2.1 剥离石墨烯的表征
图1为NMP剥离石墨烯的透射电镜(TEM)照片,可观察到透明的、薄的纳米片,这表明石墨粉已被NMP成功地剥离成石墨烯。此外,用扫描电镜(SEM)对玻碳电极以及剥离石墨烯修饰玻碳电极的表面形貌进行了分析,结果如图2所示。图2A为未修饰的玻碳电极,电极表面光滑,无显著特征;图2B为修饰剥离石墨烯的玻碳电极,其表面被纳米片所覆盖,表面富有层次;修饰的石墨烯纳米片将明显增加玻碳电极的响应面积和富集能力。

图1 剥离石墨烯的透射电镜(TEM)图Fig.1 TEM image of liquid exfoliated graphene

图2 玻碳电极(A)和修饰玻碳电极(B)的扫描电镜(SEM)图Fig.2 SEM images of bare GCE(A) and modified GCE(B)
2.2 剥离石墨烯的增敏效应

图3 空白溶液在GCE(a)和修饰GCE(c)表面的微分脉冲伏安曲线;50 nmol/L阿昔洛韦溶液在GCE(b)和修饰GCE(d)表面的微分脉冲伏安曲线Fig.3 DPV curves of bare GCE (a) and modified GCE (c) in the blank buffer solution;DPV curves of bare GCE (b) and modified GCE (d) in the presence of 50 nmol/L acyclovir Buffer solution,pH=5.0 acetate buffer;accumulation time,3 min;amount of graphene suspension,5.0 μL.
在pH=5.0的乙酸缓冲溶液中,用微分脉冲伏安法(DPV)比较了阿昔洛韦在玻碳电极和剥离石墨烯修饰玻碳电极上的氧化行为,结果如图3。不存在阿昔洛韦时,玻碳电极(曲线a)和石墨烯修饰玻碳电极(曲线c)上未出现任何氧化峰的响应,说明图3中其他曲线的氧化峰是由阿昔洛韦引起的;在未修饰玻碳电极表面,50 nmol/L的阿昔洛韦在0.94 V处出现相对较低的一个氧化峰(曲线b),说明阿昔洛韦在裸玻碳电极表面的氧化活性低;在剥离石墨烯修饰玻碳电极上,阿昔洛韦的氧化峰显著增强(曲线d),表明剥离石墨烯纳米片对阿昔洛韦的氧化有十分明显的增强效应。
从扫描电镜图可以发现,由于修饰电极明显提高了玻碳电极的表面粗糙度,从而为阿昔洛韦的氧化提供了更多的位点和更大的面积,导致阿昔洛韦在石墨烯修饰电极上的氧化信号显著提高。总之,NMP剥离石墨烯对阿昔洛韦的氧化表现出更高的活性,极大地提高了阿昔洛韦的氧化峰电流和检测灵敏度。
2.3 电化学检测参数优化
2.3.1缓冲溶液pH值的优化比较了50 nmol/L阿昔洛韦在不同pH值的乙酸缓冲溶液中的氧化峰电流,结果见图4。当pH从3.6增加到5.0时,阿昔洛韦在剥离石墨烯修饰电极表面的氧化峰电流逐渐增加;pH值继续增加到6.0时,阿昔洛韦的氧化峰电流逐渐下降。因此选用pH=5.0的0.1 mol/L的乙酸缓冲溶液。
2.3.2NMP剥离石墨烯悬浮液用量的优化如图5所示,当剥离石墨烯修饰量从0增加到5.0 μL时,阿昔洛韦的氧化峰电流显著增加,继续增加剥离石墨烯的量到10.0 μL时,阿昔洛韦的氧化峰电流变化不大,这表明电极表面的剥离石墨烯趋于稳定。综合考虑检测灵敏度和制备修饰电极的时间,剥离石墨烯的修饰量选择5.0 μL。
2.3.3富集时间的优化比较了50 nmol/L阿昔洛韦在不同富集时间下的氧化峰电流。当富集时间从0延长到3 min时,阿昔洛韦的氧化峰电流显著增加,表明富集可以明显提高检测的灵敏度。继续增加富集时间,阿昔洛韦的氧化峰电流增加缓慢,这说明电极表面阿昔洛韦的量趋于极限值。因此,测定时富集时间选择3 min。

图4 pH值对阿昔洛韦氧化峰电流的影响Fig.4 The influence of pH value on the oxidation current of acyclovir Buffer solution,pH=5.0 acetate buffer;accumulation time,3 min;amount of graphene suspension,5.0 μL.

图5 剥离石墨烯用量对阿昔洛韦氧化峰电流的影响Fig.5 The influence of amount of graphene suspension on the oxidation current of acyclovir Buffer solution,pH=5.0 acetate buffer;accumulation time,3 min.
2.3.4电极重现性与干扰物质排除实验中修饰电极每次测定后,重复测定阿昔洛韦时响应信号明显降低,这可能是由于阿昔洛韦在电极表面强的吸附所致。为此,每支修饰电极只测定1次,并考察了多支修饰电极之间的重现性,11支修饰电极平行测定50 nmol/L阿昔洛韦的相对标准偏差(RSD)为4.7%,这表明修饰电极的制备和测定都有较好的重现性。
此外还研究了其它物质对阿昔洛韦测定的干扰,发现样品可能含有的物质:0.1 mmol/L的葡萄糖、甘氨酸和色氨酸,0.05 mmol/L的抗坏血酸、尿酸、多巴胺、半胱氨酸、胱氨酸和酪氨酸,0.01 mmol/L的黄嘌呤和次黄嘌呤均不干扰50 nmol/L阿昔洛韦的氧化信号(峰电流改变<10%)。
2.4 方法的分析性能
在优化条件下,考察了该方法的线性范围和检出限。当阿昔洛韦的浓度(c)从2.5 nmol/L增加到300 nmol/L时,其氧化峰电流(Ipa,μA)随浓度线性增加,回归方程为:Ipa=0.0439c,相关系数为0.996,基于3倍信噪比(S/N=3)计算检出限为1.0 nmol/L。与已有报道的其他方法相比,本方法具有更低的检出限,见表1。
2.5 分析应用
方法被用于阿昔洛韦注射液和片剂实际样品的检测,阿昔洛韦注射液用超纯水稀释2 000倍后直接测定;片剂研磨成粉末,精确称取0.1 g溶于50 mL的0.1%NaOH溶液,超声30 min,过滤洗涤后,于250 mL容量瓶定容。取50.0 μL样品如1.2.2方法检测。同时用HPLC方法[21]分析样品,结果如表2。

表1 本方法与其他分析方法的对比Table 1 Comparison of this method and other methods
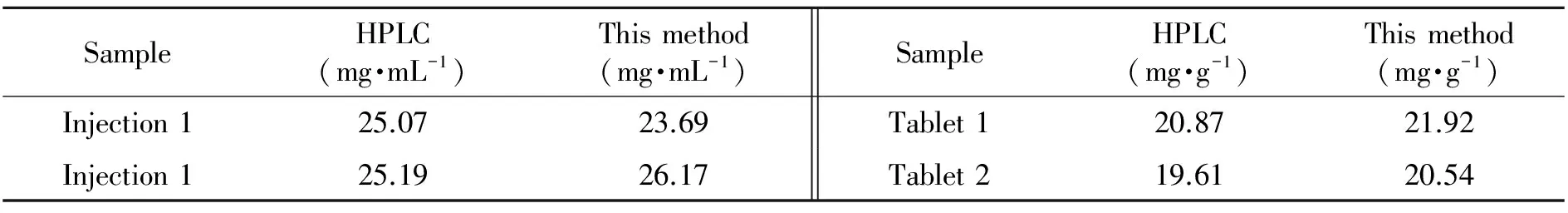
表2 HPLC方法与电化学方法分析结果对比Table 2 Comparison of HPLC and electrochemical method
3 结论
方法以NMP为溶剂,通过超声剥离制得石墨烯纳米片,剥离石墨烯由于大的表面积和强的富集能力,显著地提高了阿昔洛韦的氧化信号。在优化各项参数的基础上,建立了一种测定阿昔洛韦的新电化学方法,方法灵敏度高、准确性高、实用性强,实际样品检测结果与HPLC方法一致。
参考文献:
[1] Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A.Science,2004,306(5696):666.
[2] Zhou M,Zhai Y M,Dong S J.Analytical Chemistry,2009,81(14):5603.
[3] Brownson D A C,Kampouris D K,Banks C E.Chemical Society Reviews,2012,41(21):6944.
[4] Wu C,Sun D,Li Q,Wu K B.Sensors and Actuators B:Chemical,2012,168(12):178.
[5] Zhang B,Fan L X,Zhong H W,Liu Y W,Chen S L.Journal of the American Chemical Society,2013,135(27):10073.
[6] Gan T,Sun J Y,Wu Q,Jing Q,Yu S.Electroanalysis,2013,25(6):1505.
[7] Hernandez Y,Nicolosi V,Lotya M,Blighe F M,Sun Z Y,De S,Mcgovern I T,Holland B,Byrne M,Gun’ko Y K,Boland J J,Niraj P,Duesberg G,Krishnamurthy S,Goodhue R,Hutchison J,Scardaci V,Ferrari A C,Coleman N.Nature Nanotechnology,2008,3(9):563.
[8] Sharma M,Nautiyal P,Jain S,Jain D.Journal of AOAC International,2010,93(5):1462.
[9] Wang N N,Tang Y H,Xiong X Y,Han X N,Yu C L.Analytical Letters,2006,39(5):973.
[10] Wang F,Chen L,Chen X X,Hu S H.Analytica Chimica Acta,2006,576(1):17.
[11] Joseph R,Kumar K G.Analytical Sciences,2011,27:67.
[12] Heli H,Zarghan M,Jabbari A,Parsaei A,Moosavi-Movahedi A A.Journal of Solid State Electrochemistry,2010,14(5):787.
[13] Wang P,Gan T,Zhang J F,Luo J,Zhang S H.Journal of Molecular Liquids,2013,177(1):129.
[14] Shetti N P,Malode S J,Nandibewoor S T.Bioelectrochemistry,2012,88:76.
[15] Wang F,Chen L,Chen X,Hu S.Analytica Chimica Acta,2006,576(1):17.
[16] LIU Q Y,HE X Y,YANG T,JIA J.Journal of Analytical Science (刘琼燕,何晓英,杨涛,贾晶.分析科学学报):2014,30(3):342.
[17] QIN Z H,SHEN G M.Journal of Analytical Science (秦宗会,沈光明.分析科学学报):2008,24(3):319.
[18] Long X,Chen F.Luminescence,2012,27(6):478.
[19] Dao Y J,Jiao Z,Zhong M K.Journal of Chromatography B:Analytical Technologies in the Biomedical & Life Sciences,2008,867(2):270.
[20] Lv J,Luo L,Zhang Z.Analytica Chimica Acta,2004,510(1):35.
[21] Brown S D,White C A,Chu C K,Bartlett M G.Journal of Chromatography B,2002,772(2):327.
猜你喜欢
广州化工(2023年15期)2024-01-04 13:36:48
中国资源综合利用(2017年2期)2018-01-22 02:44:59
中西医结合心血管病电子杂志(2016年22期)2017-03-03 22:56:11
中国实用医药(2016年29期)2016-12-26 14:04:49
中国实用医药(2016年27期)2016-11-30 12:55:05
分析测试学报(2015年5期)2016-01-13 06:18:50
池州学院学报(2015年3期)2016-01-05 01:13:13
西南军医(2015年5期)2015-01-23 01:24:42
中华皮肤科杂志(2014年3期)2014-12-19 12:54:55
中国药业(2014年21期)2014-05-26 08:56:52
