
Quantum Dynamics of Oxyhydrogen Complex-Forming Reactions for the HO2and HO3Systems
2018-05-07 02:04JunxiangZuoXixiHuDaiqianXie
Jun-xiang ZuoXi-xi HuDai-qian Xie
Institute of Theoretical and Computational Chemistry,Key Laboratory of Mesoscopic Chemistry,School of Chemistry and Chemical Engineering,Nanjing University,Nanjing 210093,China
I.INTRODUCTION
Molecular reaction dynamics,which focuses on the fundamental law and nature of the chemical reaction at the level of atom and molecule,is one of the most important branches of theoretical and computational chemistry.Due to the quantum mechanical nature of the motion of electron and nucleus,only quantum treatments can provide the definitive characterisation of the reaction dynamics.The quantum dynamical theories have achieved great success during the past 40 years[1–11]since the first quantum state-resolved dynamical calculation for the simplest H+H2system was carried out[1].Quantum state-resolved dynamical study can provide the most detailed microscopic mechanism for chemical reactions and has become a major tool for understanding the multi-dimensional dynamics,tunneling,isotopic effect,mode selectivity,nonadiabatic effect,and reaction resonance state.The close collaboration between theory and experiment has uncovered some important dynamical characteristics for many important elementary reactions,such as F+H2/HD[6,8,12],Cl+H2/HD[10,13],and HD+OH[14].
Bimolecular reactions accompanied by the formation of reaction intermediates have attracted much attention due to their crucial role in combustion,atmosphere,and interstellar media.These reactions are usually barrierless in the entrance channel and involve at least one potential well along the reaction path,which are called as complex-forming reactions.The kinetic and dynamic characteristics of complex-forming reactions are much different from those in direct reaction which has only one potential barrier.Experiments can provide a lot of valuable information but cannot present very clear and complete understanding of the detailed microscopic mechanism for these reactions.So,the accurate theoretical studies are highly demanded.Both quasi classical trajectory(QCT)and quantum dynamic methods can provide detailed information on reaction dynamics,such as cross sections,product state distributions,and rate constants.The QCT method is less expensive but cannot describe the quantum effects correctly.Accurate quantum dynamical calculations for complexforming reaction are still difficult,because of the large basises or grids required for converged quantum description in potential well(s)and long-range electrostatic interaction range[15].

FIG.1 Schematic illustration of the reaction pathways for(a)HO2and(b)HO3reaction systems.The energies are taken from the results of Li et al.[92]in eV relative to the ground state reactant asymptotes for HO2and the results from the calulations by using MRCI-F12 method with VDZ-F12 basis in eV relative to the global minimum for HO3,respectively.
There actions O+HO2→HO+O2andO+OH→H+O2are elementary reaction processes that govern HOxradical concentration in the upper stratosphere[16]and mesosphere[17,18]and play significant roles in the cycle for ozone depletion.The O+HO2is also a chain-breaking step in combustion processes.Substantial experimental and theoretical interest have been attracted focusing on the kinetic and dynamic characteristics of these reactions.However,HO2and HO3are still regarded as dynamically “elusive”,presenting as yet unresolved challenges for rigorous quantum theoretical treatment.In this review,we give a detailed presentation in the advance of kinetic and dynamic investigations of these complex-forming reactions and attempt to reveal some potential characteristics of complex-forming reaction.
II.THE HO2SYSTEM
The endothermic reaction between hydrogen atom(H)and oxygen molecule(O2)

is long known as the most important combustion reaction[19]because of its crucial role in chain breaking of hydrogen oxidation and molecular oxygen consuming step.Such reaction also represents an important prototype complex-forming reaction with deep well(2.32 eV,as shown in FIG.1(a)).The reverse reaction betweenand O(3P)also plays an important role in atmospheric chemistry[17]as well as in interstellar chemistry[20].
To understand the mechanism of such reactions,considerable attentions have been attracted in theoretical studies on the adiabatic potential energy surface(PES).Since Melius and Blint[21]constructed the first analytic PES(MB PES)of the system in 1979,PES for the ground electronic stateof HO2has been developed by several groups at differentab initiolevels of theory.Earlier PESs include the double many-body expansion(DMBE)IV PES[22]which combinedab initioand experimental data simultaneously,the diatomicsin-molecules(DIM)surface[23]which utilized the diatomics in molecules model to fitab initiodata,and an analytical representation[24](TU PES)of anab initiopotential focusing on the minimum energy path(MEP)and the anisotropies in its vicinity.Among the aforementioned PESs,DMBE IV PES is probably the most popular and widely used in dynamical studies for the reasonable behavior in describing global properties of the reaction.Besides,several deficiencies of the semi-empirical PES have been reported[24–30],which may signi ficantly affect the accuracy of kinetics.For instance,the reaction was suggested to process a direct reaction channel by the dynamic study on the DMBE IV PES[27],which conflicts with the experimental complex-forming mechanism.To better understand the vibrational spectrum and dynamics of the system,Xuet al.[31,32]recently calculated about 18000ab initiopoints using multi-reference con figuration interaction method with Davidson correction and an augmented quadruple zeta correlation consistent basis set(MRCI+Q/AVQZ).They constructed a new global PES(XXZLG PES)ofby three-dimensional cubic spline interpolation.
Due to the nature of complex-forming reaction with a deep well,most theoretical dynamics studies ofreaction have been carried out by using QCT[33–39]or statistical models[31,40–43].Serious deficiencies have been pointed out,despite some reasonable results have been obtained by the heroic efforts.Dynamic bottleneck in energy transfer of the system[15]was proposed due to the observations of many back trajectories violating zero point energy(ZPE)of the OH product[35]and non-statistical limit of the H+O2reaction[37].The statistical approach was found to reproduce only a few total reaction probabilities and led to serious discrepances with exact total reactive integral cross-sections(ICS)[42–44].Several observations have con firmed that the H+O2reaction is not completely statistical[39,44–46].The dynamics of the reverse OH+O reaction are also extensively investigated utilizing QCT method[22,24,38,47–54].Like in the H+O2reaction,it is also not completely perfect when QCT approach was used in treating the dynamics of the OH+O reaction and dynamic bottleneck still exists in spite of using different PESs[36–38].Therefore,a quantum treatment is very necessary.It is still a controversial issue that whether or not the OH+O reaction is statistical.The signi ficant non-reactive scattering was found in earlier QCT studies[22,38,47–50],implying that the reaction has a non-statistical component,while latter work revealed that the non-statistical channel is unimportant[24].Some evidences have manifested that the origin of the non-statistical behavior could be attributed to the relative short living HO2complex and the inefficient intramolecular vibrational energy redistribution(IVR)[29,30,43,44,54,55].
The quantum mechanical characterization of the HO2system has been reported by many groups on various PESs since the end of last century.TheJ=0 total reaction probability was reported by Packet al.[56,57]using time-independent quantum mechanical(TIQM)method on DMBE PES at several collision energies and a threshold was found,which is consistent with the endothermicity of the H+O2reaction.It was confirmed later by quantum dynamical studies[55,58,59].Numerous sharp resonances were shown in total reaction probability[55,58–61],revealing the complex for ming mechanism.TIQM calculations on H+O2reaction are extended to total angular momentumJ>0 and Coriolis coupling was explicitly included[27,62–64],which revealed that the importance of Coriolis coupling increases withJincreasing.Due to its extensive Coriolis coupling,HO2system served as a rigorous benchmark for helicity conserving method andJ-shifting models[65–68].Poirieret al.[69–71]addressed approximated quantum dynamics calculations on the HO2bound rovibrational state using various theorybasedJ-shifting schemes.They found that the rovibrational energy levels were extremely well performed even up to largeJvalues where Coriolis coupling cannot be ignored by so-called “modified Effective Potential”(modEP)method[71].
The XXZLG PES has been widely used in quantum dynamical studies.Both time-dependent quantum mechanical(TDQM)and TIQM methods were used to calculate the total and state-to-state quantum mechanical reaction probability.Remarkably dynamic difference was found,comparing with the results on DMBE PES,especially at collision energies larger than 1.2 eV[59].The reactivity of the H+O2reaction was significantly enhanced by the effect of the vibrational excitation[72],as expected in terms of the Polanyi rule[73].The initial rotational excitation on the O2reactant was later carried out by using Chebyshev wave packet calculations and the higher rotational states of O2seem to bring better agreement with experiments[74].The quantum rate constants[72,74]were found to be significantly improved on the XXZLG PES compared with those obtained on the DMBE PES,and the temperature dependence of Arrihenius type was reasonably produced.Even though,the calculated values still underestimated compared with the experimental values and the influence factors were not ascertained.Regarding to the reaction,the quantum rate constants[75]on the XXZLG PES were qualitatively consistent with a low-temperature extrapolation of earlier experimental values but smaller than the most recent experiments at the lowest temperatures reported by Cartyet al.[45],which may stem from the particularly vulnerable region of the channel with difficulty in the long-range interaction fitted by splines[32,76,77].It has been demonstrated[15,78,79]that the long-range force plays an important role at low-temperature.Two reproducing kernel Hilbert space(RKHS)PESs[80]were designed in order to better elucidate the dynamics and accurately describe the potential well corresponding to the bound state of HO2.
To remedy the deficiencies in the fitting,especially at long-range interaction region,of XXZLG PES,Varandaset al.[81]have very recently developed a new PES for HO2(X2A′′),commonly known as Combined-Hyperbolic-Inverse-Power-Representation(CHIPR)PES,which employed the slightly correctedab initiodata of Xuet al.[32]using the novel Varandas CHIPR method[82].The main topographical features of CHIPR PES are in good agreement with XXZLG PES,as shown in FIG.2.However,some differences in long-range region,such as the undulations visible of XXZLG PES,may lead to distinguished behavior in the OH+O dynamics.Rate constants calculated by QCT method on this new PES are in general agreement with the XXZLG results from moderate temperatures upwards,but the disparity can be up to 25%at low temperatures[81].It is thus con firmed that long-range forces in the barrierless OH+O reaction could dominate the rate constants[83,84].Time-independent quantum scattering calculations have been performed on the CHIPR PES and thermal rate coefficients are reported,as shown in FIG.3,by using theJ-shifting approximation[85].It is a remarkable fact that the most recent experimental values of Cartyet al.[45]showed a distinct trend from other early experiments but close to the calculated ones.The quantum rate constants considering the influence of quantum effects in the intermediate complex were somewhat larger than the QCT results[81],although a similar pattern was reproduced.Meanwhile,the hydrogen isotopic substitution(X=Mu,H,D and T)reactions were performed by employing time-independent method and the effect of isotopic substitution on state specified rate constants was investigated[86].Rate constants of the reactions with H,D,and T presented quite similar behavior while the Mu+O2rate constants were smaller than others over the entire temperature range,especially at low temperature.Reaction probabilities for different initial rovibrational states of the OH radical were reported on both CHIPR and DMBE PESs using 3D time-dependent quantum wave-packet method[87].As is visible from the reaction probabilities forJ=0,the reactivity of OH(v=0,J=0−5)+O reaction does not always increase with the rotational excitation of OH.As presented in FIG.3,all theoretical rate constants have a negative temperature dependence over∼50 K and deviate substantially from the experiments of Cartyet al.[45]at lower temperature.It may conclude thatJ-shifting method can only be utilized to determine thermal rate coefficients correctly.Full quantum mechanical calculations with largerJvalues are required for low temperatures.However,it is still difficult for the deep well reaction with large number of coupled states.

FIG.2 Topographical features of CHIPR(a)and XXZLG(b)PESs for an oxygen atom moving around a partially relaxed OH such as to cover the range between the optimum value at the saddle point and the one at the asymptote.Adapted with permission from Ref.[81](Copyright 2013 American Institute of Physics).
The first excited electronic state ofis degenerated with its ground state ofat linear geometries and correlated to the same product of OH+O,which is associated with the reaction between the H atom and the singlet molecular oxygen molecule,

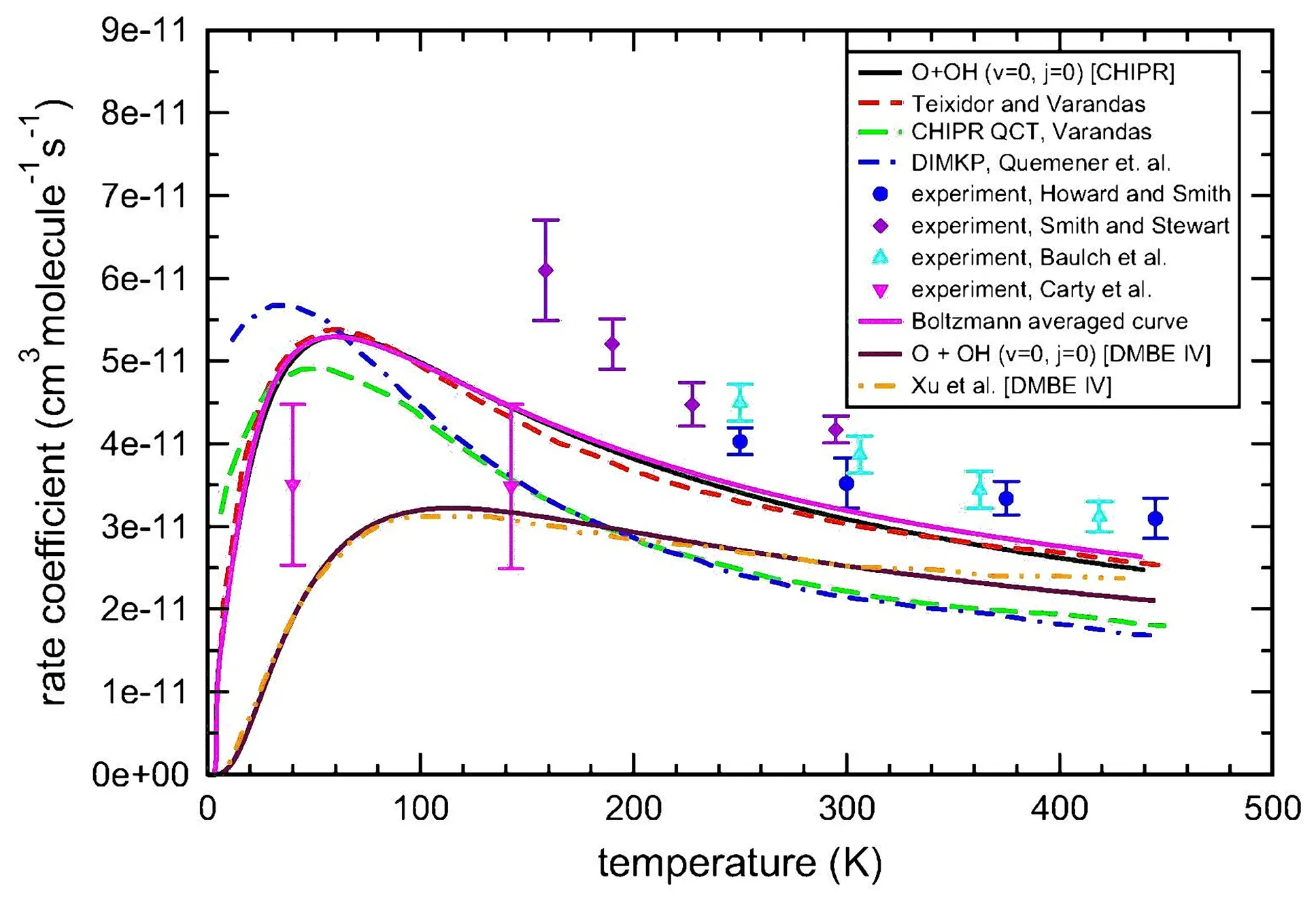
FIG.3 Comparison of the theoretical rate coefficients for O+OH→H+O2reaction.Adapted with permission from Ref.[87](Copyright 2017 Elsevier B.V.).
TABLE I Recommended rate constants for product channels of reaction.

TABLE I Recommended rate constants for product channels of reaction.
also exists.The investigation of intersystem crossing between the second doublet PES and the lowest quartet PES is thus necessary,which should provide the possibility of considering the nonadiabatic transition between theand
Furthermore,at high pressures,third-body collision is important in the following recombination reaction[96],

where Mis a third molecule.This reaction can lead to the formation of the active complexand stabilize it subsequently,making the reaction a chain-termination step.Indeed,represents all the low energy resonances occurring in the scattering process of
Despite the long history of the research in thereaction[46,92,94–101],the intrinsic mechanism of the reaction is still ambiguous. Recently,Chukalovskyet al. [102]carried out a comprehensive analysis of all available data evaluated from experiments,modeling,and theoretical estimations on the rate constants of possible channels.They argued that the non-adiabatic transition of the HO2complex from the first excited state(2A′′)to the ground quartet state(4A′),assumed by Sharipov and Starik[95],was unable to interpret the discrepancy between experimental and theoretical rate constants ofand the high probability for reaction channel(3).It was known that the lowest doublet electronic states(2A′′)and(2A′)of HO2are degenerated in collinear geometries and rovibronically coupling leads to Renner-Teller effect[15,103].The Renner-Teller coupling between ground state(2A′′)and first excited state(2A′)of HO2provides the probability of opening an additional dissociation channel of the excitedcomplex via atomic H and triplet molecularformation,which was suggested to account for the mechanism of thereaction[102].It revealed that the IVR process of excited HO2(2A′)is crucial in product channels.Ab initiocalculations,taking into account both the lowest doublet states,may provide detailed information on both reaction and quenching channels.However,no such calculations have been performed yet.Thus,further extensiveab initiostudy is highly desirable.Based on those available data,the temperature dependence ofreaction rate constants was reproduced and the approximate Arrhenius expressions of the reactions were recommended(see Table I)[102]. The activation energy of thechannel,3040 K,was close to the entrance energy barrier,3017 K(0.26 eV),of recent HO2(2A′)PES[92].In addition,the pressure dependence of thereaction was investigated by shock tube experiments[102],and the recommended expression is also shown in Table I.
Besides,three conical intersections exist between the ground stateand the excited statePESs of HO2,including one occurring at C2vgeometry(that is,O−H−O)and the other two occur at linear geometries(that is,H−O−O and O−O−H).And it was found that the HO2PES around C2vconical intersection lies lower in energy and could be easily encircled by low energy trajectories.The geometric phase(GP)effect[104,105],also known as Berry’s phase[106],stemming from the sign change of the Born-Oppenheimer adiabatic electronic wave function when the nuclei encircle a close path around a conical intersection,was shown to control the outcome of an ultracold(<1 mK)chemical reaction in the HO2system.Kendricket al.[107–112]have performed numerous theoretical studies on GP effect by taking H+O2reaction as a prototype.In order to include GP effect in three-dimension quantum scattering calculations,Kendrick and Pack[107]have presented a general vector potential approach in hyperspherical coordinates and a hybrid discrete variable representation and finite basis representation(DVR/FBR)numerical technique for C2vconical intersection in HO2.Subsequently,these methods were applied to include GP effect in H+O2scattering calculations for zero total angular momentum[108].The results of signi ficant shift in the resonance spectrum and significant changes in state-to-state transition probabilities manifest explicitly the key role of GP effect.These differences clearly demonstrated that GP effect should be crucial for H+O2scattering calculations and cannot be ignored even at low collision energy.Bound state spectrum of HO2was also calculated with and without GP effect[109].However,GP effect makes no difference in the vibrational energies even for extremely highlying states near dissociation.To further understand the mechanism and the rotationally and vibrationally resolved reaction rates in ultracold chemistry due to GP effect,scattering calculations of the O+OH(v=0,j=0)→H+O2(v′,j′)reaction forJ=0−5 have been carried out very recently[111,112].They draw an interference mechanism that the GP effect arises from the interference of the two scattering amplitudes between the direct and looping pathways.Both rotationally resolved reaction rates and vibrationally ones can be enhanced or suppressed nearly two orders of magnitude when the GP effect is included in calculations,as shown in Table II.For instance,reaction rate of 2.1×10−14cm3/s forj′=3 at 1 µK with GP is about sixty times smaller than the corresponding one without GP(1.2×10−12cm3/s),while the GP result(8.4×10−13cm3/s)is over forty times larger than that without the GP(2.0×10−14cm3/s)forj′=1.The external electric field was demonstrated to significantly modulate the GP effect,and it offers a probability that the GP effect can switch on or off the reactivity and play a role of“quantum switch” owning to the changed sign of the interference term[111].Confusingly,the number of bound state structure in electric field does not switch between extreme values,+1 and−1,as expected.The real mechanism for Stark effect on GP effects still remains elusive.A more detailed investigation on GP effect with external electric field is highly demanded.
III.THE HO3SYSTEM
The hydrogen trioxy radical(HO3)has been known as an important participant in the combustion[113–117],atmosphere chemistry[118–125],and biological processes[126–131].It is also a very important open shell species in the atmosphere,formed by adduct of OH and O2.In organic chemistry,HO3has been implicated as an intermediate in ozonation reactions[116,132].Two exothermic reactions

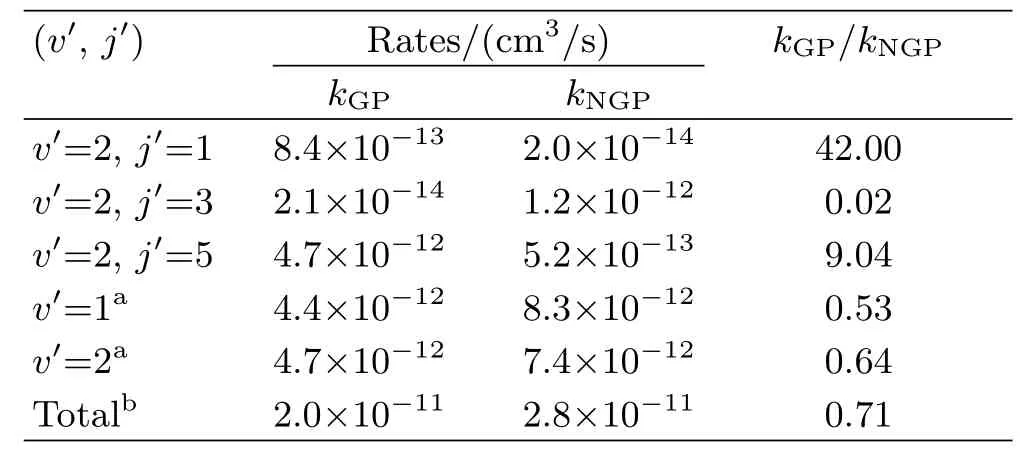
TABLE II Ultracold(1µK)rates forO+OH(v=0,j=0)→H+O2(v′,j′)reaction with(GP)and without(NGP)geometric phase effect.Data are extracted from Ref.[112].
The HO3radical was first detected by Cacaceet al.using neutralization-reionization mass spectrometry[133].Its IR spectra have been reported in argon matrices[134]and in irradiated H2O/O2ice mixtures[120,135].Rotational transitions fortrans-HO3andtrans-DO3in the gas phase were observed by Sumaet al.using Fourier-transform microwave spectroscopy,but thecisones were not observed[136].Based on the derived rotational constants and theoretical calculations of dipole moments,they concluded that thetransplanar isomer is a weakly bound molecule with a fairly long central O−O bond.In 2008,Derroet al.[137]measured the fundamental frequencies for each vibrational mode of HO3and DO3in the gas phase using IR-UV double resonance technique.The observed frequencies are significantly different from previous experimental and theoretical results.Harmonic vibrational frequencies have been calculated by Zhouet al.[138]using the explicitly correlated RCCSD(T)-F12,RS2(LS/IPEA),MRCI+Q,and MR-AQCC methods. However,the an harmonic effect is hard to be conclusive due to highly demanding inab initiocalculations.Braams and Yu[139]have done rigorous ro-vibrational calculations,based on a local PES calculated at HCTH/aug-cc-pVTZ level.Unfortunately,the vibrational energy levels show large discrepancies with experimental values[119,137],due to the inaccuracy of the PES.To do the force field calculations,hundreds of points at the vicinity of thetrans-andcis-HO3minima were calculated at MRCI+Q/AVQZ level by Sumaet al.[140].The calculated vibrational frequencies well reproduce the available experimental values,as shown in Table III.The elusive HO3radical,which is a very difficult system forab initiocharacterization,has been characterized systematically using various benchmarkab initiomethods,as reviewed in Ref.[141,142].However,there remain significant discrepancies between experiment and theory with respectto its structural properties and stability,as summarized in Table IV.For example,the bond length of the central O−O bond ranges from 1.4 ˚A to 1.7 ˚A by differentab initiomethods[117,122,136,139,143–151],while the experimental value is 1.688˚A[136]or 1.684˚A[152].Numerous high-levelab initioresults suggested that multi-reference methods predicted longer central O−O bond length.Besides,the relative stability betweentrans-andcis-HO3is different with different theoretical method.Some earlyab initiotheoretical work and DFT studies predicted a non-planar structure,while recent calculations reached consensus that HO3intermediate is planar with bothtransandcisgeometries.In 2011,Varandas[147]explored the MEP for isomerization of HO3using high-levelab initiomethods with extrapolation to the complete basis set limit,as shown in FIG.4.The results showed that,for single reference methods,thecis-HO3is slightly more stable than thetrans-HO3.In turn,the multi-reference calculations predicted that thetrans-HO3is more stable,which is in agreement with the available experimental evidence.

TABLE III Fundamental frequencies of HO3obtained from several experimental and theoretical studies.
In addition,there has been much debate on the binding energy of HO3,as shown in Table IV.An upper limit of the dissociation energy was estimated to be 5.31 or 6.12 kcal/mol from experiments using infrared action spectroscopy[118,119,137,153].In 2010,Le Picardet al.[154]derived a value of(2.9±0.07)kcal/mol on the basis of experimental kinetic studies,in which the OH concentration was measured in the presence of excess O2using laser-induced fluorescence.A number of theoretical investigations at different levels of theory and basis set predicted 4−10 kcal/mol for the binding energy of HO3[139,148,150,155,156].Most recently,Varandas[151]calculated the dissociation energiesDe,which are 4.5 and 4.7 kcal/mol for thecisandtransisomers,respectively.TheD0value fortrans-HO3is in good agreement with the commonly accepted value(2.9±0.07)kcal/mol from the low-temperature CRESU experiment.FIG.5 shows the dissociation curves for the reaction HO3→HO+O2at different levels of theory.The EOMIP-CCSD curve[148]predicted a significant barrier which is about 5 kcal/mol above thetransminimum and then drops steeply to reach the asymptote at 2.5 kcal/mol.The existence of such a barrierconflicts with the experimental evidence,in which a strong negative temperature dependence[154]for the reversed association reaction suggests a barrier-free reaction.This observation suggests that the commonly used single-reference CCSD approach might not be able to provide an accurate global PES,due apparently to the multi-reference characters of the system.
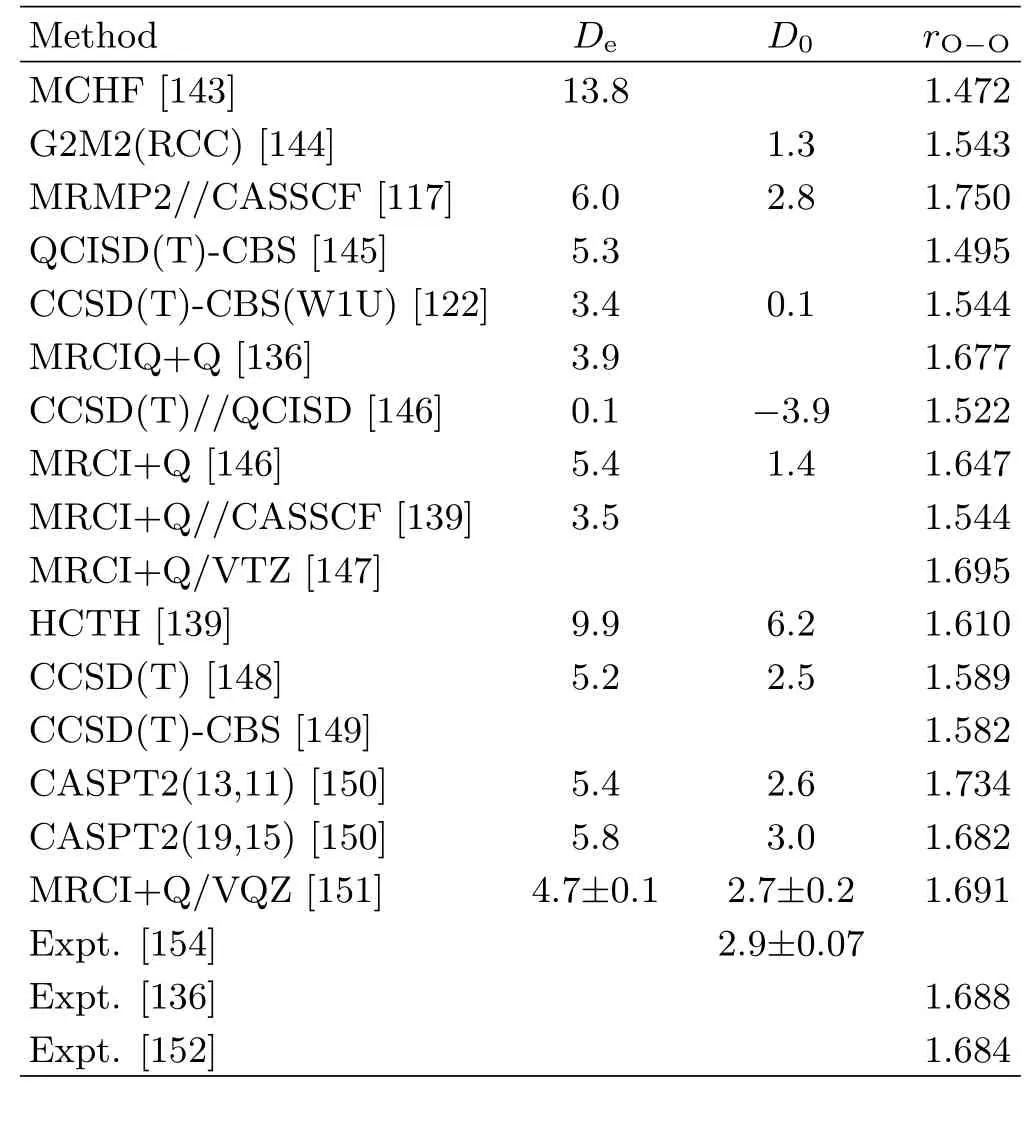
TABLE IV A comparison of classical dissociation energies(Dein kcal/mol)for HO3→OH+O2,dissociation energies at 0 K(D0in kcal/mol)and central O−O bond lengths(rO−Oin ˚A)for trans-HO3from different theoretical and experimental studies.
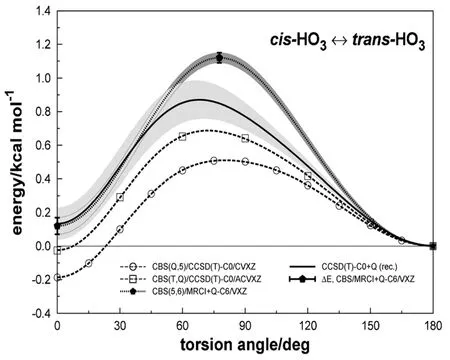
FIG.4The isomerization path of Adapted with permission from Ref.[147](Copyright 2011 Royal Society of Chemistry).
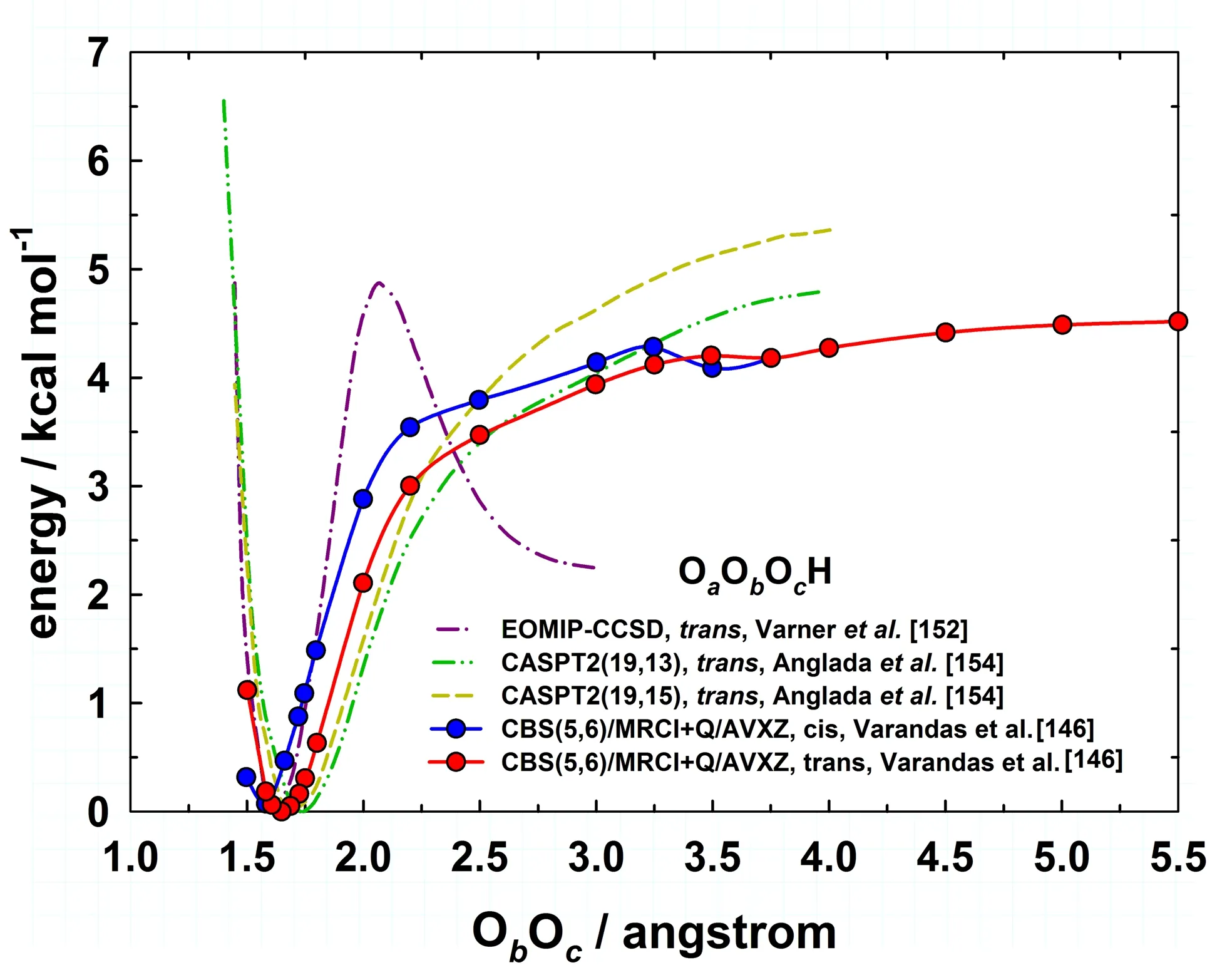
FIG.5 Dissociation curves for the reaction as it progresses from the trans-and cis-HO3geometries(energies taken relative to the trans-HO3minimum).
The first global PES for the ground state HO3(DMBE I)was constructed by Varandas and Yu[157]using UCISD method.The PES predicts a metastable HO3structure,which conflicts with the experimental evidence and high-levelab initiocalculations.In 2001,Varandaset al.[145]obtained a new PES for this system using the QCISD(T)/CBS method,named DMBE II.This PES has a dissociation barrier,which is inconsistent with the available experimental evidence,and two stable planar HO3isomers,in which thecis-HO3is more stable than thetransone.These PESs are not sufficiently accurate due to the low level ofab initiotheory and to the relatively small number ofab initiopoints.Another analytical PES in the many body expansion form was obtained by fitting to about 28000 HCTH/aug-cc-pVTZ points[139].The PES predicts aD0of 6.15 kcal/mol,and implies a central O−O bond length of 1.610˚A fortrans-HO3,which is shorter than the commonly accepted value of 1.688˚A.And the vibrational frequencies carried out on this PES have large discrepancies with the experimental results[119,137].
Several kinetic measurements have been conducted to determine the rate constants for the O+HO2reaction[158–162],which can be used to simulate the concentrations of OH,HO2,and O3in the upper stratosphere and lower mesosphere. The reported rate coefficients of this exothermic reaction have a negative temperature dependence at low temperatures. The measured rate constant for the reaction O+HO2→HO+O2was found to be independent with N2pressure from 10 torr to 500 torr with a mean value of(6.2±1.1)×10−11cm3/(molecule·s)at 298 K by Ravishankaraet al.[162],which is in good agreement with the value of(6.1±0.4)×10−11cm3/(molecule·s)reported by Keyser[158]. Bruneet al. [160]and Sridharanet al.[163]reported that the rate constants for this reaction at 300 K are(5.2±0.8)×10−11and∼5.7×10−11cm3/(molecule·s),respectively.In 1987,Nicovichet al.[161]employed a pulsed laser photolysis technique to investigate the kinetic of this reaction over the range of 266−391 K at 80 torr of N2.Their results followed the Arrhenius expression:k(T)=(2.91±0.7)×10−11exp[(228±75)/T]cm3/(molecule·s).The rate constant recommended by the NASA kinetic data evaluation panel is an average of these five studies.However,the atmosphere chemistry reaction model using the recommended value for this reaction underestimated the ozone concentration in the mesosphere[17,18].Based on the DMBE I PES,the rate constant,product distributions,and differencial cross sections for the reaction O+HO2→HO+O2by employing the QCT method[164]and a reduced three-dimensional quantum dynamic method[165]showed large differences between themselves and the available experimental data.In the reduced-dimensional quantum mechanical study,the reaction was treated as coplanar and the in finite-order sudden approximation was applied,which gave too low values of rate constants and cross sections. In 2004,Silveiraet al.[166]found that the vibrational excitation has minor effect on the dynamics based on the QCT calculations on the DMBE I and DMBE II PESs,respectively.In addition to the complex-forming pathway,this reaction can also proceed via a direct hydrogen abstraction pathway on two doublet(2A′′and2A′)and two quadruplet(4A′′and4A′)states.The abstraction barriers are approximately 2−4 kcal/mol based onab initiocalculations[117,143].As a result,this pathway will become viable at high temperatures and contribute to the rate constant.
Neither the complex-forming nor abstraction pathway was accurately described by the existing HO3PESs.To better understand the kinetics and dynamics of the relevant reactions,we suggested to map out new accurate global PESs of the ground and excited electronic states for this system using the MRCI-F12 approach and the permutation invariant polynomial neural network(PIP-NN) fitting approach[167].The spin orbital couplings should also be included.The PESs will include the OH+O2,O+HO2,and H+O3channels,as well as the complex-forming and direct abstraction pathways.The PESs will greatly advance our under-standing of the HO3system and associated reactions.We are particularly keen on obtaining accurate theoretical rate constants for the related reactions,which can be used to reduce uncertainties in combustion and atmosphere kinetic models.In addition,a full-dimensional quantum mechanical characterization for this system has not been accomplished due to the involvement of three heavy atoms and the deep potential wells.Further theoretical improvements on decreasing the number of basis and improving computational efficiency in quantum dynamic method are very desirable.
IV.SUMMARY
In this review,we have presented an overview of the recent progresses in two prototypical oxyhydrogen complex-forming reactions of the HO2and HO3systems.A number of achievements have been obtained from the unremitting efforts of numerous researchers,whereas there are still many questions to be illuminated.For HO2,several global PESs for the ground electronic state have been constructed,and the predicted rate constants are in reasonable agreement with the available observed values,while some major discrepancies still exist.The nonadiabatic effect has been found to be crucial in the HO2system and nonadiabatic quantum dynamical calculations with excited PESs,including electronic states of 12A′′,12A′,22A′′and4A′′,are necessary.In particular,the influence of the GP effect on the reaction dynamics ofneeds to be further addressed.Besides,different PESs predicted disparity state-to-state dynamics for the reaction of H+O2,especially at higher collision energies,so that molecular beam experiment is very demanding to con firm the detailed reaction mechanism.For HO3,only the minimum energy path on the ground electronic state has been widely investigated,but no reliable reaction dynamics for the related reactions are performed due to the absence of an accurate global PES and to the difficulty of carrying out quantum dynamics for such complex-forming reactions.
Thanks to the rapid development of the computing method and program of quantum chemistry,it is now feasible to perform high levelab initiocalculation using MRCI-F12 for the electronic ground and excited states for the HO2and HO3systems.The artificial neural network(NN)method,which has been found to be the most efficient approach to fitting high dimensional PES[168],can be applied to generate the global PES for the ground and excited electronic states.Based on the accurate PESs,it is possible to perform the detailed quantum dynamics by employing the efficient wave packet propagation method.Although such dynamics calculations are still hard to implement for complex-forming reactions with a deep well,such as the reactions related to the HO3system,the computational efficiency could be remarkably improved by using the optimized coordinates and suitable absorption in the boundary.With the close interplay between theory and experiment,it is anticipated that the detailed reaction dynamics for the reactions related to the HO2and HO3systems could be revealed in the near further.
V.ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China(No.91641104,No.21733006,and No.21590802).
[1]G.C.Schatz and A.Kuppermann,J.Chem.Phys.65,4642(1976).
[2]G.C.Schatz,Annu.Rev.Phys.Chem.39,317(1988).
[3]S.C.Althorpe and D.C.Clary,Annu.Rev.Phys.Chem.54,493(2003).
[4]S.C.Althorpe,F.Fernándezalonso,B.D.Bean,J.D.Ayers,A.E.Pomerantz,R.N.Zare,and E.Wrede,Nature416,67(2002).
[5]S.A.Harich,D.X.Dai,C.C.Wang,X.M.Yang,S.D.Chao,and R.T.Skodje,Nature419,281(2002).
[6]M.Qiu,Z.Ren,L.Che,D.Dai,S.A.Harich,X.Wang,X.Yang,C.Xu,D.Xie,and M.Gustafsson,Science311,1440(2006).
[7]D.C.Clary,Science321,789(2008).
[8]W.Dong,C.Xiao,T.Wang,D.Dai,X.Yang,and D.H.Zhang,Science327,1501(2010).
[9]Z.Sun,L.Liu,S.Y.Lin,R.Schinke,H.Guo,and D.H.Zhang,Proc.Nat.Acad.Sci.USA107,555(2010).
[10]T.Yang,J.Chen,L.Huang,T.Wang,C.Xiao,Z.Sun,D.Dai,X.Yang,and D.H.Zhang,Science347,60(2015).
[11]X.Hu,L.Zhou,and D.Xie,WIRES Comput.Mol.Sci.8,e1350(2018).
[12]T.Yang,L.Huang,Y.Xie,T.Wang,C.Xiao,Z.Sun,D.Dai,M.Chen,D.H.Zhang,and X.Yang,Chin.J.Chem.Phys.28,471(2015).
[13]X.Wang,W.Dong,C.Xiao,L.Che,Z.Ren,D.Dai,X.Wang,P.Casavecchia,X.Yang,B.Jiang,D.Xie,Z.Sun,S.Y.Lee,D.H.Zhang,H.J.Werner,and M.H.Alexander,Science322,573(2008).
[14]C.Xiao,X.Xu,S.Liu,T.Wang,W.Dong,T.Yang,Z.Sun,D.Dai,X.Xu,D.H.Zhang,and X.Yang,Science333,440(2011).
[15]H.Guo,Int.Rev.Phys.Chem.31,1(2012).
[16]K.W.Jucks,D.G.Johnson,K.V.Chance,W.A.Traub,J.J.Margitan,G.B.Osterman,R.J.Salawitch,and Y.Sasano,Geophys.Res.Lett.25,3935(1998).
[17]M.E.Summers,R.R.Conway,D.E.Siskind,M.H.Stevens,D.Offermann,M.Riese,P.Preusse,D.F.Strobel,and J.M.Russell,Science277,1967(1997).
[18]P.Crutzen,Science277,1951(1997).
[19]J.A.Miller,R.J.K.And,and C.K.Westbrook,Annu.Rev.Phys.Chem.41,345(1990).
[20]I.W.M.Smith,E.Herbst,and Q.Chang,Mon.Not.R.Astron.Soc.350,323(2004).
[21]C.F.Melius and R.J.Blint,Chem.Phys.Lett.64,183(1979).
[22]M.R.Pastrana,L.A.M.Quintales,J.Brandao,and A.J.C.Varandas,J.Phys.Chem.92,4552(1990).
[23]B.Kendrick and R.T.Pack,J.Chem.Phys.102,1994(1995).
[24]J.Troe and V.G.Ushakov,J.Chem.Phys.115,3621(2001).
[25]A.J.Dobbyn,M.Stumpf,H.M.Keller,and R.Schinke,J.Chem.Phys.103,9947(1995).
[26]R.Schinke,H.M.Keller,H.Flothmann,M.Stumpf,C.Beck,D.H.Mordaunt,A.J.Dobbyn,S.A.Rice,R.A.Marcus,J.Troe,D.M.Neumark,and M.E.Kellman,inChemical Reactions and Their Control on the Femtosecond Time Scale Xxth Solvay Conference on Chemistry,P.Gaspard and I.Burghardt Eds.,745(1997).
[27]A.J.H.M.Meijer and E.M.Gold field,J.Chem.Phys.108,5404(1998).
[28]L.B.Harding,A.I.Maergoiz,J.Troe,and V.G.Ushakov,J.Chem.Phys.113,11019(2000).
[29]S.Y.Lin,D.Xie,and H.Guo,J.Chem.Phys.125,091103(2006).
[30]C.Xu,B.Jiang,D.Xie,S.C.Farantos,S.Y.Lin,and H.Guo,J.Phys.Chem.A111,10353(2007).
[31]C.Xu,D.Xie,D.H.Zhang,S.Y.Lin,and H.Guo,J.Chem.Phys.122,244305(2005).
[32]C.Xu,D.Xie,P.Honvault,S.Y.Lin,and H.Guo,J.Chem.Phys.127,024304(2007).
[33]J.A.Miller,J.Chem.Phys.74,5120(1981).
[34]A.J.C.Varandas,J.Brandão,and M.R.Pastrana,J.Chem.Phys.96,5137(1992).
[35]A.J.C.Varandas,J.Chem.Phys.99,1076(1993).
[36]C.Y.Yang and S.J.Klippenstein,J.Chem.Phys.103,7287(1995).
[37]J.A.Miller and B.C.Garrett,Int.J.Chem.Kinet.29,275(1997).
[38]J.A.Miller and S.J.Klippenstein,Int.J.Chem.Kinet.31,753(1999).
[39]G.Lendvay,D.Xie,and H.Guo,Chem.Phys.349,181(2008).
[40]S.Y.Lin,E.J.Rackham,and H.Guo,J.Phys.Chem.A110,1534(2006).
[41]T.González-Lezana,Int.Rev.Phys.Chem.26,29(2007).
[42]P.Bargueno,T.Gonzalez-Lezana,P.Larrégaray,L.Bonnet,and J.Claude Rayez,Phys.Chem.Chem.Phys.9,1127(2007).
[43]P.Bargueño,T.González-Lezana,P.Larrégaray,L.Bonnet,J.C.Rayez,M.Hankel,S.C.Smith,and A.J.H.M.Meijer,J.Chem.Phys.128,244308(2008).
[44]Z.Sun,D.H.Zhang,C.Xu,S.Zhou,D.Xie,G.Lendvay,S.Y.Lee,S.Y.Lin,and H.Guo,J.Am.Chem.Soc.130,14962(2008).
[45]D.Carty,A.Goddard,S.P.K.Köhler,I.R.Sims,and I.W.M.Smith,J.Phys.Chem.A110,3101(2006).
[46]J.Ma,H.Guo,C.Xie,A.Li,and D.Xie,Phys.Chem.Chem.Phys.13,8407(2011).
[47]J.A.Miller,J.Chem.Phys.84,6170(1986).
[48]L.A.M.Quintales,A.J.C.Varandas,and J.M.Alvarino,J.Phys.Chem.92,4552(1988).
[49]J.Davidsson and G.Nyman,J.Chem.Phys.92,2407(1990).
[50]G.Nyman and J.Davidsson,J.Chem.Phys.92,2415(1990).
[51]A.I.Maergoiz,E.E.Nikitin,J.Troe,and V.G.Ushakov,J.Chem.Phys.108,5265(1998).
[52]L.B.Harding,J.Troe,and V.G.Ushakov,Phys.Chem.Chem.Phys.2,631(2000).
[53]M.Jor fi,P.Honvault,P.Halvick,S.Y.Lin,and H.Guo,Chem.Phys.Lett.462,53(2008).
[54]M.Jor fi,P.Honvault,P.Bargueño,T.González-Lezana,P.Larrégaray,L.Bonnet,and P.Halvick,J.Chem.Phys.130,184301(2009).
[55]P.Honvault,S.Y.Lin,D.Xie,and H.Guo,J.Phys.Chem.A111,5349(2007).
[56]R.T.Pack,E.A.Butcher,and G.A.Parker,J.Chem.Phys.99,9310(1993).
[57]R.T.Pack,E.A.Butcher,and G.A.Parker,J.Chem.Phys.102,5998(1995).
[58]D.H.Zhang and J.Z.H.Zhang,J.Chem.Phys.101,3671(1994).
[59]S.Y.Lin,H.Guo,P.Honvault,and D.Xie,J.Phys.Chem.B110,23641(2006).
[60]M.Hankel,S.C.Smith,and A.J.H.M.Meijer,J.Chem.Phys.127,064316(2007).
[61]G.Quéméner,B.K.Kendrick,and N.Balakrishnan,J.Chem.Phys.132,014302(2010).
[62]A.J.H.M.Meijer and E.M.Gold field,J.Chem.Phys.110,870(1999).
[63]E.M.Gold field and A.J.H.M.Meijer,J.Chem.Phys.113,11055(2000).
[64]A.J.H.M.Meijer and E.M.Gold field,Phys.Chem.Chem.Phys.3,2811(2001).
[65]H.Zhang and S.C.Smith,J.Chem.Phys.118,10042(2003).
[66]H.Zhang and S.C.Smith,J.Chem.Phys.120,9583(2004).
[67]H.Zhang and S.C.Smith,J.Chem.Phys.123,014308(2005).
[68]H.Zhang,and S.C.Smith,J.Phys.Chem.A110,3246(2006).
[69]W.Chen and B.Poirier,J.Theor.Comput.Chem.09,435(2010).
[70]C.Petty,W.Chen,and B.Poirier,J.Phys.Chem.A117,7280(2013).
[71]C.Petty and B.Poirier,Chem.Phys.Lett.605-606,16(2014).
[72]S.Y.Lin,Z.Sun,H.Guo,D.H.Zhang,P.Honvault,D.Xie,and S.Y.Lee,J.Phys.Chem.A112,602(2008).
[73]M.G.Evans,and M.Polanyi,Trans.Faraday Soc.35,178(1939).
[74]S.Y.Lin,H.Guo,G.Lendvay,and D.Xie,Phys.Chem.Chem.Phys.11,4715(2009).
[75]F.Lique,M.Jor fi,P.Honvault,P.Halvick,S.Y.Lin,H.Guo,D.Q.Xie,P.J.Dagdigian,J.K Klos,and M.H.Alexander,J.Chem.Phys.131,221104(2009).
[76]G.Quéméner,N.Balakrishnan,and B.K.Kendrick,Phys.Rev.A79,022703(2009).
[77]J.C.Juanes-Marcos,G.Quemener,B.K.Kendrick,and N.Balakrishnan,Phys.Chem.Chem.Phys.13,19067(2011).
[78]J.M.C.Marques and A.J.C.Varandas,Phys.Chem.Chem.Phys.3,505(2001).
[79]P.F.Weck and N.Balakrishnan,Int.Rev.Phys.Chem.25,283(2006).
[80]D.Xie,C.Xu,T.S.Ho,H.Rabitz,G.Lendvay,S.Y.Lin,and H.Guo,J.Chem.Phys.126,074315(2007).
[81]A.J.C.Varandas,J.Chem.Phys.138,134117(2013).
[82]A.J.C.Varandas,J.Chem.Phys.138,054120(2013).
[83]D.C.Clary and H.J.Werner,Chem.Phys.Lett.112,346(1984).
[84]D.C.Clary and J.P.Henshaw,Faraday Discuss.Chem.Soc.84,333(1987).
[85]M.M.Teixidor and A.J.C.Varandas,J.Chem.Phys.142,014309(2015).
[86]M.M.Teixidor and A.J.C.Varandas,Chem.Phys.Lett.638,61(2015).
[87]S.Ghosh,R.Sharma,S.Adhikari,and A.J.C.Varandas,Chem.Phys.Lett.675,85(2017).
[88]H.Du and J.P.Hessler,J.Chem.Phys.96,1077(1992).
[89]K.Keßler and K.Kleinermanns,J.Chem.Phys.97,374(1992).
[90]P.Jensen,R.J.Buenker,J.P.Gu,G.Osmann,and P.R.Bunker,Can.J.Phys.79,641(2001).
[91]J.A.K Klos,F.Lique,M.H.Alexander,and P.J.Dagdigian,J.Chem.Phys.129,064306(2008).
[92]A.Li,D.Xie,R.Dawes,A.W.Jasper,J.Ma,and H.Guo,J.Chem.Phys.133,144306(2010).
[93]J.Ma,S.Y.Lin,H.Guo,Z.Sun,D.H.Zhang,and D.Xie,J.Chem.Phys.133,054302(2010).
[94]P.Szabó and G.Lendvay,J.Phys.Chem.A119,7180(2015).
[95]A.S.Sharipov and A.M.Starik,Phys.Scr.88,058305(2013).
[96]A.Starik and A.Sharipov,Phys.Chem.Chem.Phys.13,16424(2011).
[97]R.L.Brown,J.Geophys.Res.75,3935(1970).
[98]C.Schmidt and H.I.Schiff,Chem.Phys.Lett.23,339(1973).
[99]L.T.Cupitt,G.A.Takacs,and G.P.Glass,Int.J.Chem.Kinet.14,487(1982).
[100]W.Hack and H.Kurzke,J.Phys.Chem.90,1900(1986).
[101]P.Szabó and G.Lendvay,J.Phys.Chem.A119,12485(2015).
[102]A.A.Chukalovsky,K.S.Klopovsky,A.P.Palov,A.M.Yu,and T.V.Rakhimova,J.Phys.D:Appl.Phys.49,485202(2016).
[103]V.V.Melnikov,T.E.Odaka,P.Jensen,and T.Hirano,J.Chem.Phys.128,114316(2008).
[104]H.C.Longuethiggins,U.Opik,M.H.L.Pryce,and R.A.Sack,Proc.R.Soc.London,Ser.A244,1(1958).
[105]G.Herzberg and H.C.Longuet-Higgins,Discuss.Faraday Soc.35,77(1963).
[106]F.R.S.M.V.Berry,Proc.R.Soc.London,Ser.A392,45(1984).
[107]B.Kendrick and R.T.Pack,J.Chem.Phys.104,7475(1996).
[108]B.Kendrick and R.T.Pack,J.Chem.Phys.104,7502(1996).
[109]B.Kendrick and R.T.Pack,J.Chem.Phys.106,3519(1997).
[110]B.K.Kendrick,J.Phys.Chem.A107,6739(2003).
[111]J.Hazra,B.K.Kendrick,and N.Balakrishnan,J.Phys.Chem.A119,12291(2015).
[112]B.K.Kendrick,J.Hazra,and N.Balakrishnan,Nat.Commun.6,7918(2015).
[113]J.Yang,Q.S.Li,and S.Zhang,Phys.Chem.Chem.Phys.9,466(2007).
[114]J.P.Le Crane,M.T.Rayez,J.C.Rayez,and E.Villenave,Phys.Chem.Chem.Phys.8,2163(2006).
[115]N.K.Srinivasan,M.C.Su,J.W.Sutherland,and J.V.Michael,J.Phys.Chem.A109,7902(2005).
[116]J.Cerkovnik,E.Erzen,J.Koller,and B.Plesnicar,J.Am.Chem.Soc.124,404(2002).
[117]O.Setokuchi,M.Sato,and S.Matuzawa,J.Phys.Chem.A104,3204(2000).
[118]C.Murray,E.L.Derro,T.D.Sechler,and M.I.Lester,J.Phys.Chem.A111,4727(2007).
[119]E.L.Derro,C.Murray,T.D.Sechler,and M.I.Lester,J.Phys.Chem.A111,11592(2007).
[120]P.D.Cooper,M.H.Moore,and R.L.Hudson,J.Phys.Chem.A110,7985(2006).
[121]S.Chalmet and M.F.Ruiz-Lopez,J.Chem.Phys.124,194502(2006).
[122]W.M.F.Fabian,J.Kalcher,and R.Janoschek,Theor.Chem.Acc.114,182(2005).
[123]A.J.C.Varandas,J.Phys.Chem.A108,758(2004).
[124]H.Szichman and A.J.C.Varandas,J.Phys.Chem.A103,1967(1999).
[125]H.G.Yu and A.J.C.Varandas,J.Chem.Soc.Faraday Trans.93,2651(1997).
[126]B.Plesnicar,Acta Chimica Slovenica52,1(2005).
[127]A.N.Wu,D.Cremer,and B.Plesnicar,J.Am.Chem.Soc.125,9395(2003).
[128]X.Xu and W.A.Goddard,Proc.Nat.Acad.Sci.USA99,15308(2002).
[129]A.Engdahl and B.Nelander,Science295,482(2002).
[130]P.Wentworth,L.H.Jones,A.D.Wentworth,X.Y.Zhu,N.A.Larsen,I.A.Wilson,X.Xu,W.A.Goddard,K.D.Janda,A.Eschenmoser,and R.A.Lerner,Science293,1806(2001).
[131]S.Aloisio and J.S.Francisco,J.Am.Chem.Soc.121,8592(1999).
[132]B.Plesničar,T.Tuttle,J.Cerkovnik,J.Koller,and D.Cremer,J.Am.Chem.Soc.125,11553(2003).
[133]F.Cacace,G.de Petris,F.Pepi,and A.Troiani,Science285,81(1999).
[134]B.Nelander,A.Engdahl,and T.Svensson,Chem.Phys.Lett.339,295(2001).
[135]W.Zheng,D.Jewitt,and R.I.Kaiser,Phys.Chem.Chem.Phys.9,2556(2007).
[136]K.Suma,Y.Sumiyoshi,and Y.Endo,Science308,1885(2005).
[137]E.L.Derro,T.D.Sechler,C.Murray,and M.I.Lester,J.Chem.Phys.128,244313(2008).
[138]Y.Zhou,H.Hu,L.Li,H.Hou,and B.Wang,Comput.Theor.Chem.1026,24(2013).
[139]B.J.Braams and H.G.Yu,Phys.Chem.Chem.Phys.10,3150(2008).
[140]K.Suma,Y.Sumiyoshi,and Y.Endo,J.Chem.Phys.139,094301(2013).
[141]C.Murray,E.L.Derro,T.D.Sechler,and M.I.Lester,Acc.Chem.Res.42,419(2009).
[142]A.J.C.Varandas,Int.J.Quantum Chem.114,1327(2014).
[143]M.Dupuis,G.Fitzgerald,B.Hammond,W.A.Lester,and H.F.Schaefer,J.Chem.Phys.84,2691(1986).
[144]T.P.W.Jungkamp and J.H.Seinfeld,Chem.Phys.Lett.257,15(1996).
[145]H.G.Yu and A.J.C.Varandas,Chem.Phys.Lett.334,173(2001).
[146]A.Mansergas,J.M.Anglada,S.Olivella,M.F.Ruiz-Lopez,and M.Martins-Costa,Phys.Chem.Chem.Phys.9,5865(2007).
[147]A.J.C.Varandas,Phys.Chem.Chem.Phys.13,9796(2011).
[148]M.E.Varner,M.E.Harding,J.Vazquez,J.Gauss,and J.F.Stanton,J.Phys.Chem.A113,11238(2009).
[149]M.E.Varner,M.E.Harding,J.Gauss,and J.F.Stanton,Chem.Phys.346,53(2008).
[150]J.M.Anglada,S.Olivella,and A.Solé,J.Chem.Theory Comput.6,2743(2010).
[151]A.J.C.Varandas,J.Chem.Theory Comput.8,428(2012).
[152]M.C.McCarthy,V.Lattanzi,D.Kokkin,O.Martinez Jr.,and J.F.Stanton,J.Chem.Phys.136,034303(2012).
[153]E.L.Derro,T.D.Sechler,C.Murray,and M.I.Lester,J.Phys.Chem.A112,9269(2008).
[154]S.D.Le Picard,M.Tizniti,A.Canosa,I.R.Sims,and I.W.M.Smith,Science328,1258(2010).
[155]P.A.Denis and F.R.Ornellas,J.Phys.Chem.A113,499(2009).
[156]P.A.Denis,M.Kieninger,O.N.Ventura,R.E.Cachau,and G.H.F.Diercksen,Chem.Phys.Lett.365,Pii s0009-2614(02)01432-x 440(2002).
[157]A.J.C.Varandas and H.G.Yu,Mol.Phys.91,301(1997).
[158]L.F.Keyser,J.Phys.Chem.86,3439(1982).
[159]U.C.Sridharan,L.X.Qiu,and F.Kaufman,J.Phys.Chem.86,4569(1982).
[160]W.H.Brune,J.J.Schwab,and J.G.Anderson,J.Phys.Chem.87,4503(1983).
[161]J.M.Nicovich and P.H.Wine,J.Phys.Chem.91,5118(1987).
[162]A.R.Ravishankara,P.H.Wine,and J.M.Nicovich,J.Chem.Phys.78,6629(1983).
[163]U.C.Sridharan,F.S.Klein,and F.Kaufman,J.Chem.Phys.82,592(1985).
[164]W.Wang,R.Gonzalez-Jonte,and A.J.C.Varandas,J.Phys.Chem.A102,6935(1998).
[165]A.J.C.Varandas and H.Szichman,Chem.Phys.Lett.295,113(1998).
[166]D.M.Silveira,P.Caridade,and A.J.C.Varandas,J.Phys.Chem.A108,8721(2004).
[167]B.Jiang and H.Guo,J.Chem.Phys.139,054112(2013).
[168]Y.L.Zhang,X.Y.Zhou,and B.Jiang,Chin.J.Chem.Phys.30,727(2017).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2018年2期
CHINESE JOURNAL OF CHEMICAL PHYSICS2018年2期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Investigation on Preparation and Anti-icing Performance of Superhydrophobic Surface on Aluminum Conductor
- Study of Cadmium-Doped Zinc Oxide Nanocrystals with Composition and Size Dependent Band Gaps
- Glucose Isomerization into Fructose Catalyzed by MgO/NaY Catalyst
- Analysis of Solvent Effect on Mechanical Properties of Poly(ether ether ketone)Using Nano-indentation
- Direct Synthesis of Monodisperse Hollow Molecularly Imprinted Polymers Based on Unfunctionalized SiO2for the Recognition of Bisphenol A
- Agent-Based Network Modeling Study of Immune Responses in Progression of Ulcerative Colitis