
利用两种统计模型对中国肉用西门塔尔牛屠宰性状的全基因组关联分析
2018-05-07 08:59常天鹏夏江威宝金山金生云徐凌洋张路培李俊雅高会江
畜牧兽医学报 2018年4期
常天鹏,夏江威,宝金山,金生云,朱 波,徐凌洋,陈 燕,张路培,高 雪,李俊雅,高会江*
(1.中国农业科学院北京畜牧兽医研究所,北京 100193; 2.内蒙古锡林郭勒盟乌拉盖管理区兽医局,乌拉盖 026321)
屠宰性状是肉牛产业中最为重要的一类性状,直接影响着牛肉产量和经济效益,因此,对于屠宰性状的选育是肉牛育种者的重点研究方向。借助于现代分子生物学技术,分子标记辅助选择(MAS)显著缩短了选育周期,节省了时间和成本投入,成为当前育种工作的有效方法[1]。而实现肉牛屠宰性状MAS的关键步骤就是目标性状的数量性状基因座(QTLs)及候选基因定位[2]。目前,关于世界上主要肉牛品种屠宰性状的候选基因定位工作已经有了许多的研究报道,其中对胴体重的研究较多。A.Takasuga等[3]通过对日本黑牛进行QTLs定位,在6号染色体38 Mb和14号染色体41.7和49.8 Mb均检测到了影响胴体重的QTLs;A. K.Lindholm-Perry等[4]通过对一个包括14个品种血统的杂交牛群体进行基因定位,在6号染色体38 Mb鉴定到影响胴体重的LAP3、NCAPG、LCORL等候选基因;A.G.Doran等[5]借助于GWAS方法对荷斯坦牛的胴体性状进行分析,找到了一系列影响胴体性状的QTLs、候选基因及信号通路。
在GWAS时代到来之前,研究者主要利用QTL定位策略定位候选基因。QTL定位常用的试验群体类型有BC、F2及RIL群体等,这些群体是精心设计的,交配类型也是明确的,其目的在于使其基因型分离以增加表型的丰富性,而且还方便推测伪标记的基因型[6]。QTL定位比较经典的方法有区间定位法(IM)、复合区间定位法(CIM)[7-8]等。但是由于标记密度低,达不到精确定位的层次。随着高密度芯片技术的发展,GWAS以其较高的精确性和效率,成为当前探索复杂性状遗传机理的最有效的方法之一。GWAS选用的群体一般是自然群体,基于历史重组造成标记与QTL的连锁不平衡来定位真实QTL的位置。然而由于样本量要求大,自然群体经常会有群体分层现象,从而导致假阳性过高。目前,常用的GWAS方法有线性混合模型(LMM)和贝叶斯方法[9-10]。通过QTL定位与GWAS方法的比较,不难发现,由于QTL定位精心的试验设计,能够很好地避免GWAS存在的问题,因此先前在QTL定位积累的统计方法对GWAS研究具有很好的借鉴意义。
本试验拟尝试两种不同的统计模型:一种是线性混合模型(LMM),另一种是结合复合区间作图策略的线性混合模型(CIM-LMM),基于Illumina BovineHD (770K)芯片分型获得基因型数据,对中国肉用西门塔尔牛的胴体重和骨重性状进行关联分析,旨在筛选影响目标性状的显著SNPs并挖掘相关的功能基因;同时,比较两个方法获得的结果,为GWAS的研究方法提供新策略。
1 材料与方法
1.1 试验材料
本研究中试验动物来自中国农业科学院北京畜牧兽医研究所在内蒙古锡林郭勒盟乌拉盖管理区构建的中国肉用西门塔尔牛资源群体。该群体自2008年开始建立并记录各个阶段表型数据。为了便于数据的测量和采集,将5~9月龄的牛运送至北京金维福仁清真食品有限公司肉牛养殖场,在该场期间按照统一的饲养管理方法集中育肥,每3个月测量一次体尺和体重数据,当饲养至18~24月龄时进行分批屠宰分割。截止2016年12月底,共收集到1 301头肉牛的表型数据和基于Illuminate BovineHD (770K)芯片分型的基因型数据。
选择的目标性状为胴体重和骨重,胴体重是指牛经过宰杀放血后,除去皮、头、蹄、尾、内脏以及生殖器(母牛去除乳房)后的躯体部分重量;骨重是指胴体经过标准流程分割后剩余所有骨头的总重量。屠宰以及胴体分割流程完全按照国家标准GB/T 27643-2011进行。表1显示了胴体重和骨重两个性状的描述性统计量,包括表型数据的记录数、最小值、最大值、平均值、标准差及遗传力。
表1胴体重、骨重性状的描述性统计量
Table1Thedescriptivestatisticsofcarcassweightandboneweight
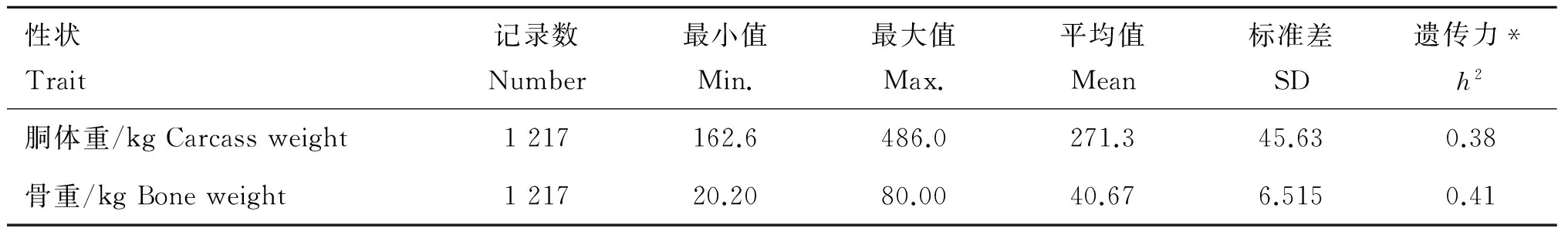
性状Trait记录数Number最小值Min.最大值Max.平均值Mean标准差SD遗传力*h2胴体重/kgCarcassweight1217162.6486.0271.345.630.38骨重/kgBoneweight121720.2080.0040.676.5150.41
*性状遗传力由R 环境下asreml包估计
*Heritabilities were estimated by asreml package in R environment
1.2 基因分型和质量控制
采集1 301个个体血样,利用TIANGEN基因组DNA提取试剂盒(天根生化科技有限公司,北京,中国)从血样中提取出基因组DNA,并利用Illumina BovineHD (770K)芯片进行基因分型。然后采用PLINK v1.07软件进行质量控制,质量控制标准为:(1)SNPs检出率>90%;(2)最小等位基因频率>10%;(3)哈代-温伯格平衡检验(P>10-6);(4)个体基因型缺失率<0.1。经过质控之后,共有1 217个个体和671 990个SNPs用于关联分析。
1.3 统计模型
利用两种统计模型:线性混合模型(LMM)和复合区间定位-线性混合模型(CIM-LMM)进行目标性状的关联分析。
LMM为GWAS最常用的统计方法:
y=Xβ+Zkγk+ξ+ε
(1)
其中,X表示固定效应系数矩阵,β是固定效应向量;Zk表示第k个SNP的指示变量,γk表示第k个SNP的效应值(作固定效应处理);ξ~N(0,Kφ2)称作微效多基因效应,K表示基于标记推断的亲缘关系矩阵,φ2表示多基因效应的方差;ε表示随机残差并且服从多元正态分布N(0,Iσ2)。该方法最早是由J.M.Yu等[10]提出,也称作常用的Q+K模型,本试验中该方法由Tassel 5.0软件中的LMM实现。
CIM-LMM的统计模型:
y=Xβ+Zk-1γk-1+Zkγk+Zk+1γk+1+ξ+ε
(2)
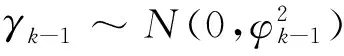
在统计检验方面,这两种方法都采用P<0.05作为筛选显著SNPs的标准。为了解决多重假设检验引起的假阳性问题,本研究采用Bonferroni法进行P值校正[12]。
1.4 群体结构分析
在进行GWAS分析时,群体结构的影响不可忽略。本研究采用主成分分析法(Principal component analysis,PCA)检测群体结构[13]。通过PCA获得基因型数据特征值和特征向量,然后选择适当的特征向量作为协变量加入关联分析统计模型中,从而实现对群体结构的校正。本研究中PCA通过R程序中的princomp函数实现。
1.5 固定效应显著性检验
固定效应的校正可以降低数据资料的不平衡性和关联分析的假阳性率。对于固定效应的分析,要把尽可能的影响因素加入到校正模型中,然后对各因素做显著性分析,将对表型值有显著影响的因素放入统计模型,不显著的影响因素去掉。本研究根据资源群体的实际情况,考虑的固定因素为出生年、季节、出生场效应,并将进场重、育肥天数、群体结构作为协变量。通过R程序中的GLM函数实现固定效应的显著性检验,其模型为y=u+year+season+farm+weight+fatten_day+pca+e,其中y为性状表型值,u为群体均值,year为出生年效应,season为出生季节效应,farm出生场效应,weight为进场重协变量,fatten_day为育肥天数协变量,pca为群体结构协变量,e为随机残差。
1.6 基因注释
利用生物信息学网站Ensemble中的BioMart模块将检测到的显著SNPs比对到牛的基因组(BostaurusUMD 3.1)中,依据SNPs的物理位置在其上下游寻找候选基因。然后通过NCBI网站的Gene数据库查找相关基因的生物学功能,并结合前人报道对候选基因进行功能注释。
2 结 果
2.1 群体结构及固定效应检验
图1为基于前两个主成分将个体聚类的群体结构图。从图中明显可以看出,个体间存在很大程度的群体分层。绝大多数个体集中于右下角区域,另外4个区域有少量个体分布。所以选取前两个主成分作为协变量来消除群体分层对关联分析的影响。在固定效应的显著性检验中,年效应、进场重及育肥天数协变量对表型值有显著影响。因此,选择将这些显著的固定效应及协变量一起放入统计模型进行关联分析。
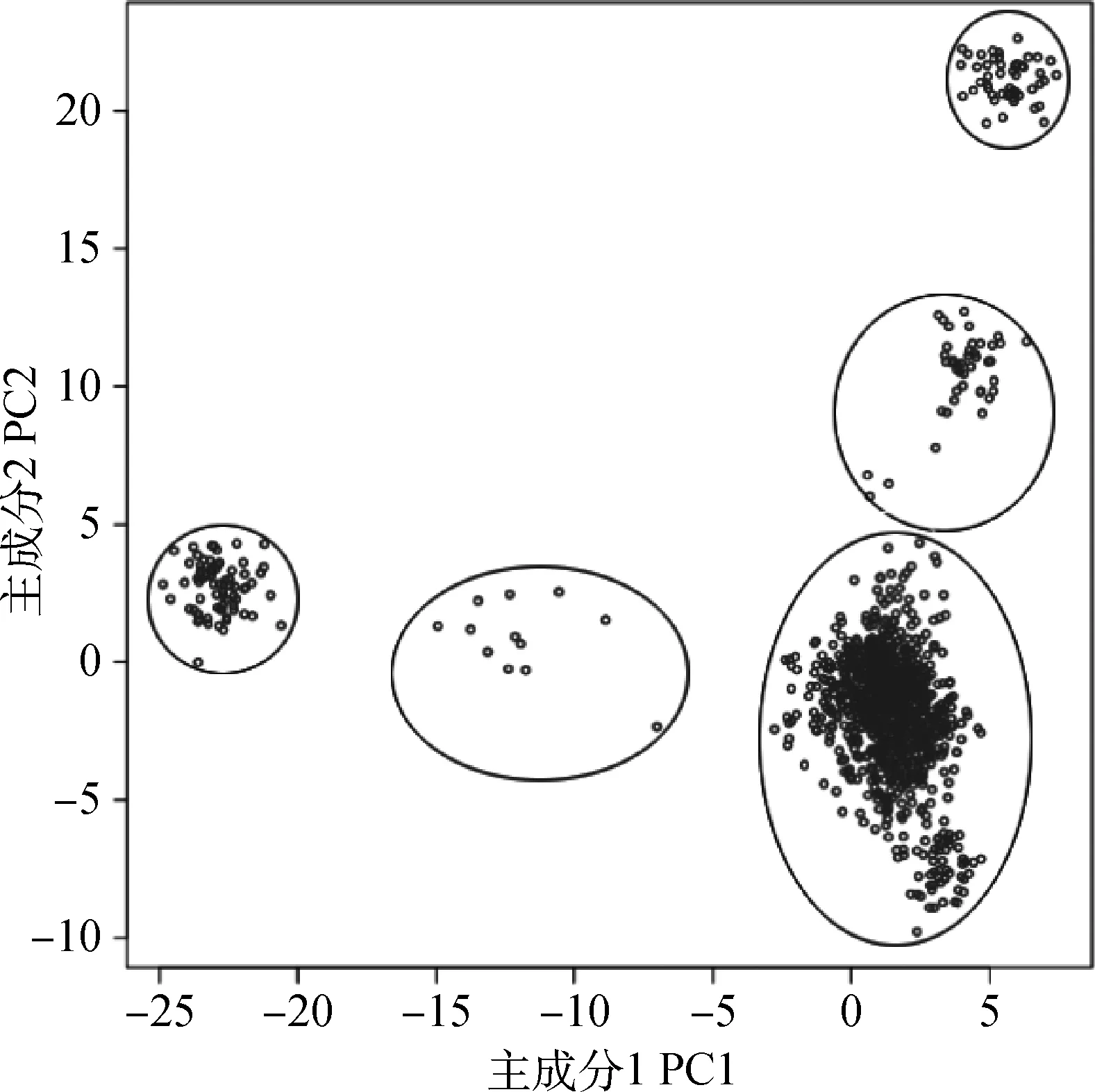
图1 根据PCA绘制群体结构图Fig.1 Population structure identified by principal components analysis
2.2 两种模型的QQ图分析
利用LMM和CIM-LMM对目标性状的GWAS结果的P值作QQ图,横坐标表示期望P值的负对数,纵坐标表示实际观察P值的负对数,见图2。可以看出,使用LMM分析得到的P值,观察值与期望值吻合度很好,大多数的点都位于对角线上,这表明此模型较好的校正了系统偏差。由CIM-LMM分析结果的得到的QQ图前端显示了一定的拖尾现象,其原因在于将目标标记k的邻近标记做随机效应处理,导致许多微效或无效标记的效应值压缩为零,而统计检验时P值接近或等于1,而后端观察值与期望值基本吻合。在统计显著性方面,CIM-LMM方法得到的-lg(P)值比LMM方法高2~5个数量级,而且识别出较多偏离期望值的SNPs。
2.3 两种模型鉴定的SNPs和候选基因
图3展示了通过LMM和CIM-LMM对目标性状GWAS分析的Manhattan图。对于这两个性状,以Bonferroni法矫正后的P值作为阈值,分别在全基因组范围内共找到8和7个显著SNPs,其中2个SNPs与胴体重及骨重都有关联。通过Ensembl网站BioMart模块鉴定了与这些SNPs密切相关的11个基因。对于胴体重,LMM方法共检测到3个显著相关的SNPs,分别位于第5、14、17号染色体上。这3个SNPs分别邻近或坐落于C12ORF74、RIMS2、BT.88981基因。CIM-LMM方法共检测到了8个显著相关的SNPs,其中包含了LMM方法识别的3个SNPs,剩余的5个SNPs分别位于第3、10、14、16号染色体上,邻近或坐落于PBX1、GCNT4、ALDH1A2、FAM84B、DUSP10基因。对于骨重,LMM方法共检测到4个显著相关的SNPs,分别位于第5、6、14号染色体上,邻近或坐落于C12ORF74、LCORL、RIMS2基因。CIM-LMM方法共找到了7个显著相关的SNPs,其中包含了LMM方法检测的4个SNPs,剩余的3个SNPs分别位于第6、14号染色体上,邻近于WDFY3、FER1L6基因。具体如表2和表3所示。
3 讨 论
3.1 群体结构的分层现象
从图1可以看出,本研究所用的中国肉用西门塔尔牛群体呈现一定的群体分层。所谓群体分层,主要由于自然选择、人工选择、遗传漂变等诸多因素导致的群体内出现了等位基因频率不同的亚群体。它会导致一些非原因等位基因同真实QTLs形成连锁不平衡而表现出与目标性状关联,从而导致伪关联或假阳性[14]。若不对群体分层加以校正,那么群体分层效应会被误认为是QTLs效应,降低关联分析准确性。因此,本研究选取PCA结果中前两个主成分作为协变量来校正群体分层,从而避免由于群体分层引起的假阳性问题。
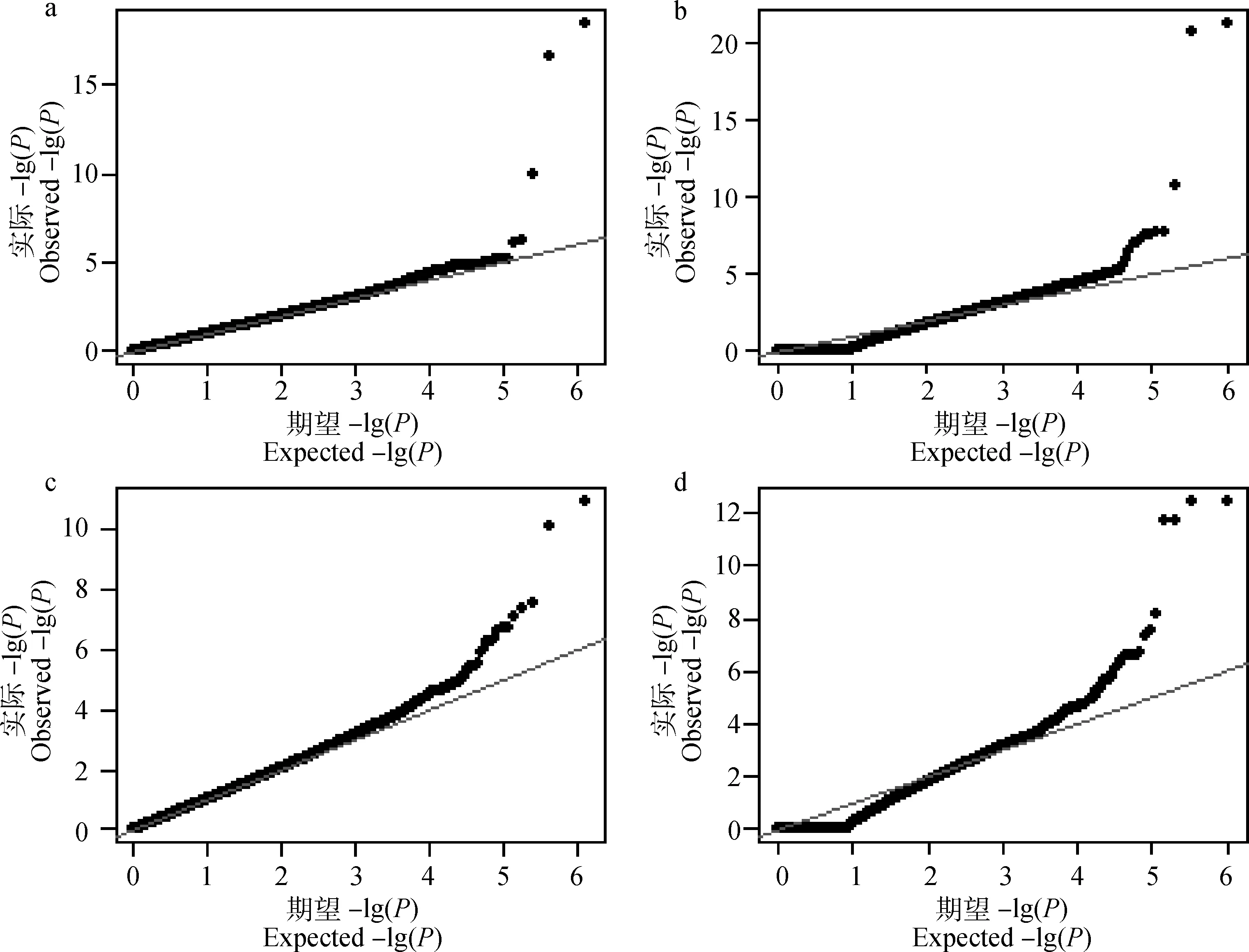
a、c.利用LMM分别对胴体重、骨重GWAS结果的QQ图;b、d.利用CIM-LMM对胴体重、骨重GWAS结果的QQ图a,c. QQ plots for the GWAS results of carcass weight and bone weight by LMM, respectively; b,d. QQ plots for the GWAS results of carcass weight and bone weight by CIM-LMM, respectively图2 两种模型对目标性状分析结果的QQ图Fig.2 QQ plots for the analyzing results of target traits by two statistic models
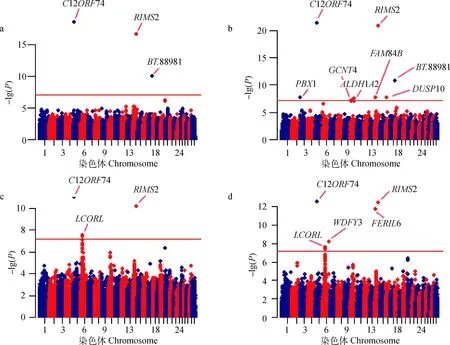
a、c. 利用LMM分别对胴体重、骨重GWAS结果作的曼哈顿图;b、d. 利用CIM-LMM对胴体重、骨重GWAS结果作的曼哈顿图a,c. Manhattan plots for the GWAS result of carcass weight and bone weight by LMM, respectively; b,d. Manhattan plots for the GWAS result of carcass weight and bone weight by CIM-LMM, respectively图3 两种模型对目标性状分析结果的曼哈顿图Fig.3 Manhattan plots for the analyzing results of target traits by two statistic models
表2两种模型对胴体重的GWAS分析结果
Table2TheGWASresultofcarcassweightbytwostatisticmodels
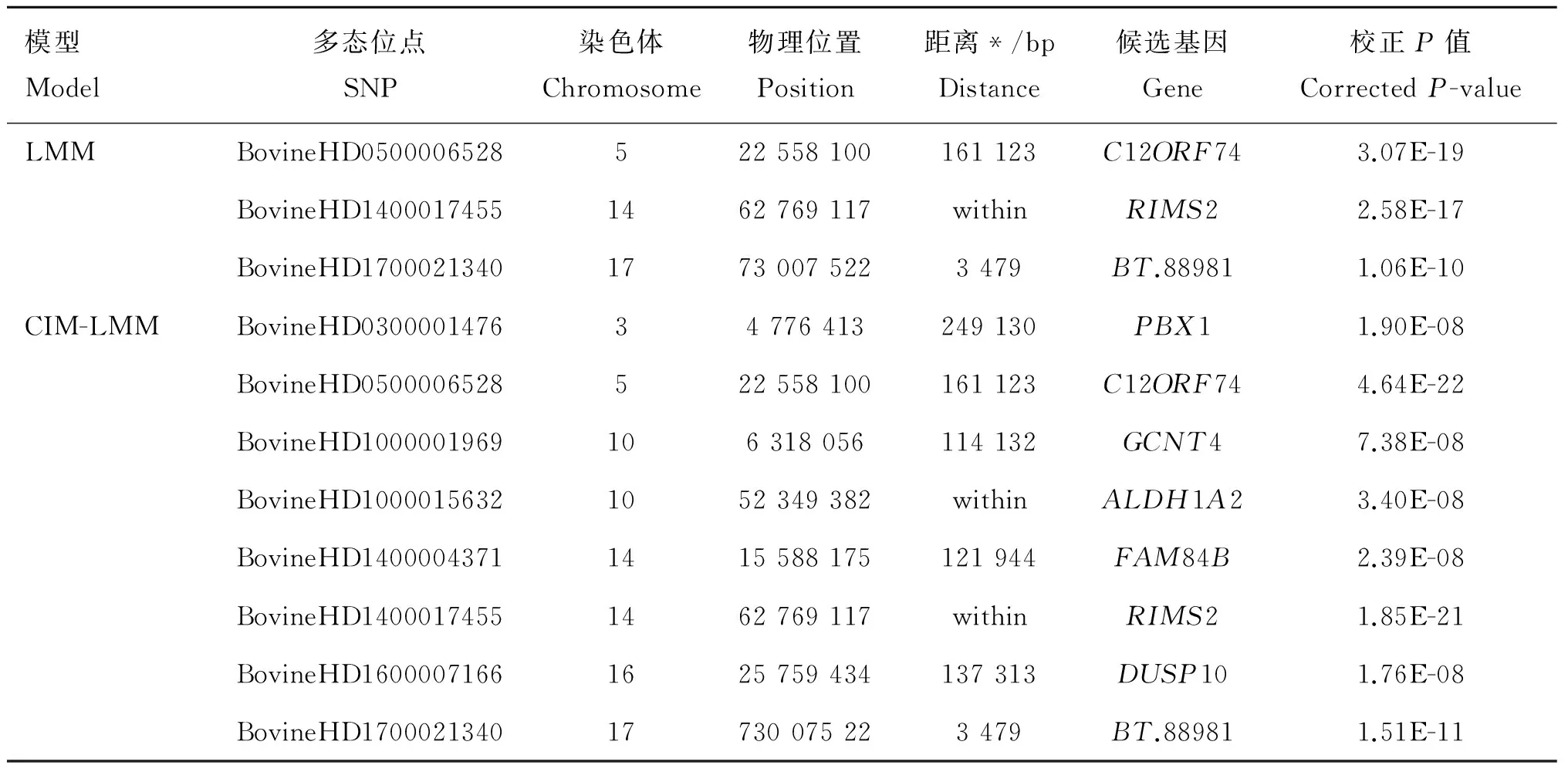
模型Model多态位点SNP染色体Chromosome物理位置Position距离*/bpDistance候选基因Gene校正P值CorrectedP-valueLMMBovineHD0500006528522558100161123C12ORF743.07E-19BovineHD14000174551462769117withinRIMS22.58E-17BovineHD170002134017730075223479BT.889811.06E-10CIM-LMMBovineHD030000147634776413249130PBX11.90E-08BovineHD0500006528522558100161123C12ORF744.64E-22BovineHD1000001969106318056114132GCNT47.38E-08BovineHD10000156321052349382withinALDH1A23.40E-08BovineHD14000043711415588175121944FAM84B2.39E-08BovineHD14000174551462769117withinRIMS21.85E-21BovineHD16000071661625759434137313DUSP101.76E-08BovineHD170002134017730075223479BT.889811.51E-11
*表示SNP与邻近基因的物理距离,单位为碱基对(bp),“within”表示落入基因内,下表同
*.Mean the physical distance between SNP and the nearest gene and the units are bp, “within” indicate that the SNP located in the nearest genes. The same as below
表3两种模型对骨重的GWAS分析结果
Table3TheGWASresultofboneweightbytwostatisticmodels
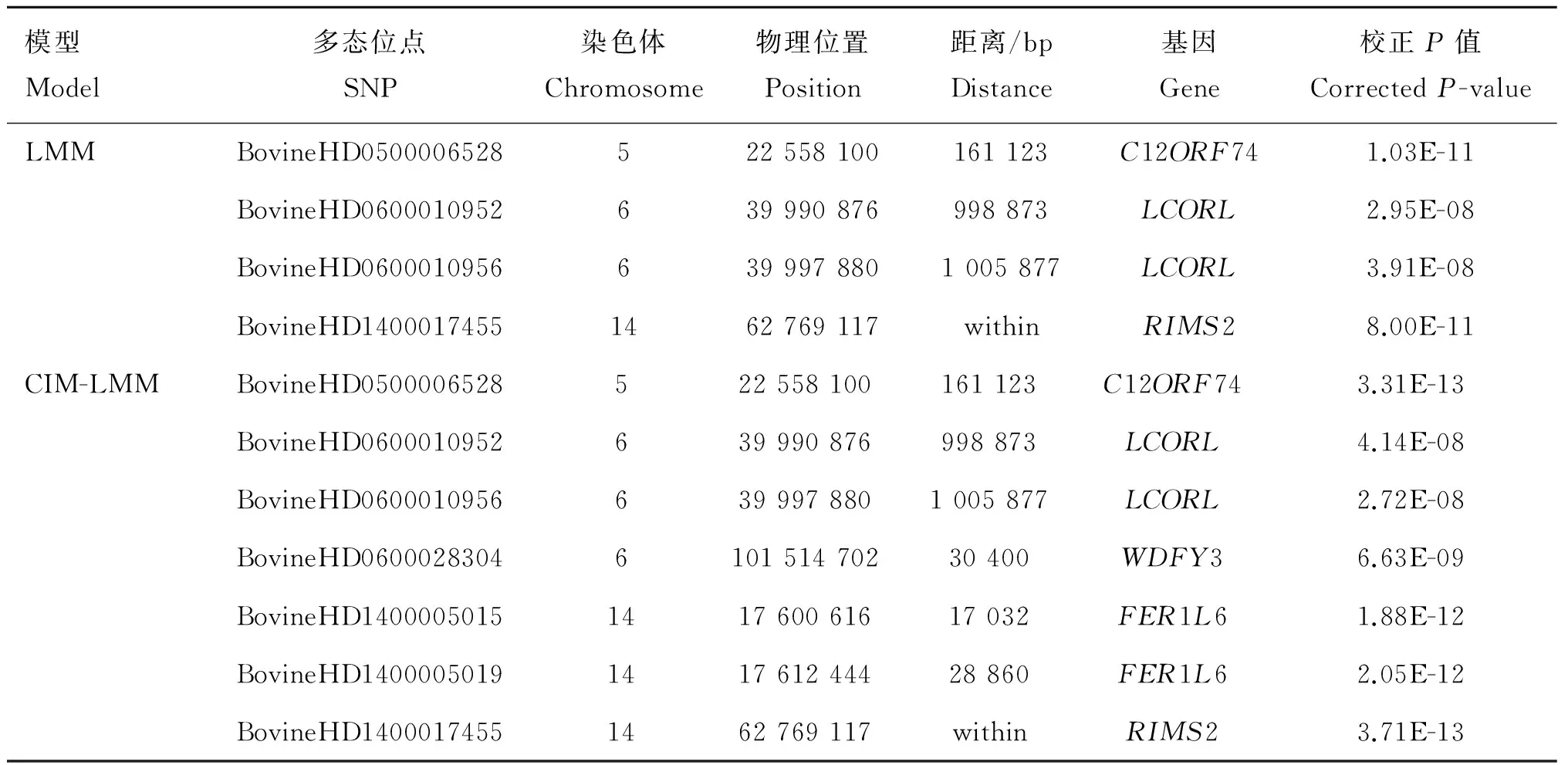
模型Model多态位点SNP染色体Chromosome物理位置Position距离/bpDistance基因Gene校正P值CorrectedP-valueLMMBovineHD0500006528522558100161123C12ORF741.03E-11BovineHD0600010952639990876998873LCORL2.95E-08BovineHD06000109566399978801005877LCORL3.91E-08BovineHD14000174551462769117withinRIMS28.00E-11CIM-LMMBovineHD0500006528522558100161123C12ORF743.31E-13BovineHD0600010952639990876998873LCORL4.14E-08BovineHD06000109566399978801005877LCORL2.72E-08BovineHD0600028304610151470230400WDFY36.63E-09BovineHD1400005015141760061617032FER1L61.88E-12BovineHD1400005019141761244428860FER1L62.05E-12BovineHD14000174551462769117withinRIMS23.71E-13
3.2 两种模型分析结果比较
两种模型GWAS结果的QQ图显示,LMM方法结果的理论值与观察值基本吻合,表明该模型较好的校正了群体分层;然而显著SNPs检测效力一般,对于两个性状,在全基因组范围内分别仅发现3、4个SNPs与之关联。CIM-LMM方法利用基于符合区间定位(CIM)的策略,在目标标记的两侧加入侧翼标记并做随机效应处理,考虑了标记之间的连锁关系同时压缩无效标记的效应;结果除了检测到LMM发现的所有SNPs,还检测到更多的SNPs,同时各SNPs的检验P值更加显著,较之于LMM方法更有优势。
3.3 候选基因的功能分析
本研究共发现11个不同的候选基因,分布于3、5、6、10、14、16、17号染色体上,且多集中于6号和14号染色体上,一些候选基因已在其他牛种或物种上被相继研究。其中,PBX1基因位于BTA3上,据报道,它与人类的骨密度有关,并且被认为是影响印第安人2型糖尿病的1个候选基因[15]。LCORL、WDFY3基因位于BTA6上,LCORL已经被证实与人类的骨骼骨架尺寸和马的肩胛骨高度有关[16],而且也被发现它是影响牛饲料摄入量和生长的调控基因[4];WDFY3已经被确定与仔猪的出生重有关,可能参与猪的胎儿发育过程[17]。GCNT4、ALDH1A2基因位于BTA10上,研究表明,牛的GCNT4基因参与脂肪酸从头合成的途径和黏液生物合成[18];ALDH1A2基因在合成视黄酸的过程中起着关键作用,视黄酸是维生素A代谢的中间产物,而维生素A又主要影响动物的身体生长和骨骼发育[19]。FAM84B、RIMS2、FER1L6基因都位于BTA14上,研究发现,FAM84B可能是影响人类食道鳞状细胞癌的候选基因[20],在牛上该基因被发现与荷斯坦公牛精子活力有关系[21];RIMS2基因是人类中枢神经系统突触膜胞外分泌的调控基因,并且与人类退化性腰脊柱侧凸(DLS)的患病风险有关[22-23];而韩牛的一项研究表明,FER1L6基因与韩牛牛肉的大理石花纹形成密切关联,并认为该基因是影响牛肉质量的候选基因[24]。
4 结 论
本研究分别使用LMM和CIM-LMM两种统计模型,对中国肉用西门塔尔牛的胴体重和骨重性状进行了全基因组关联分析。分别鉴定到8、7个显著相关的SNPs,其中有2个SNPs与这两个性状都有关联,并利用NCBI及Ensemble网站查找到影响目标性状的11个候选基因;同时还探讨了GCNT4、ALDH1A2、LCORL和WDFY3等基因的功能。此外,我们也对2种统计模型的分析结果进行比较,结果显示,对于本试验群体CIM-LMM方法较之于LMM方法具有更高的统计效力。本研究为解析中国肉用西门塔尔牛屠宰性状的遗传机理做了探索,为后期的验证研究提供了方向,也为GWAS方法研究提供了新思路。
参考文献(References):
[1] DEKKERS J C M. Commercial application of marker-and gene-assisted selection in livestock: strategies and lessons[J].JAnimSci, 2004, 82 (E-Suppl): E313-E328.
[2] DUNHAM R A. Gene mapping, quantitative trait locus mapping and marker-assisted selection[M]//DUNHAM R A. Aquaculture and Fisheries Biotechnology: Genetic Approaches. Wallingford, UK: CABI Publishing, 2004.
[3] TAKASUGA A, WATANABE T, MIZOGUCHI Y, et al. Identification of bovine QTL for growth and carcass traits in Japanese Black cattle by replication and identical-by-descent mapping[J].MammGenome, 2007, 18(2): 125-136.
[4] LINDHOLM-PERRY A K, SEXTEN A K, KUEHN L A, et al. Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle[J].BMCGenet, 2011, 12(1): 103.
[5] DORAN A G, BERRY D P, CREEVEY C J. Whole genome association study identifies regions of the bovine genome and biological pathways involved in carcass trait performance in Holstein-Friesian cattle[J].BMCGenomics,2014, 15: 837.
[6] 魏巨龙. 混合线性模型解析数量性状遗传基础的研究[D]. 北京: 中国农业大学, 2016.
WEI J L. Application of Linear Mixed Model (LMM) to dissect genetic basis of quantitative traits[D]. Beijing: China Agricultural University, 2016. (in Chinese)
[7] ZENG Z B. Precision mapping of quantitative trait loci[J].Genetics, 1994, 136(4): 1457-1468.
[8] LANDER E S, BOTSTEIN D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps[J].Genetics, 1989, 121(1): 185-199.
[9] FERNANDO R L, GARRICK D. Bayesian methods applied to GWAS[M]//GONDRO C, VAN DER WERF J, HAYES B. Genome-Wide Association Studies and Genomic Prediction. Totowa, NJ: Humana Press, 2013: 237-274.
[10] YU J M, PRESSOIR G, BRIGGS W H, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness[J].NatGenet, 2006, 38(2): 203-208.
[11] WEI J L, XU S Z. A random-model approach to QTL mapping in multiparent advanced generation intercross (MAGIC) populations[J].Genetics, 2016, 202(2): 471-486.
[12] JOHNSON R C, NELSON G W, TROYER J L, et al. Accounting for multiple comparisons in a genome-wide association study (GWAS)[J].BMCGenomics, 2010, 11: 724.
[13] PRICE A L, PATTERSON N J, PLENGE R M, et al. Principal components analysis corrects for stratification in genome-wide association studies[J].NatGenet, 2006, 38(8): 904-909.
[14] WANG D, SUN Y, STANG P, et al. Comparison of methods for correcting population stratification in a genome-wide association study of rheumatoid arthritis: principal-component analysis versus multidimensional scaling[J].BMCProc, 2009, 3(Suppl 7): S109.
[15] THAMEEM F, WOLFORD J K, BOGARDUS C, et al. Analysis ofPBX1 as a candidate gene for type 2 diabetes mellitus in Pima Indians[J].BiochimBiophysActa, 2001, 1518(1-2): 215-220.
[16] SORANZO N, RIVADENEIRA F, CHINAPPEN-HORSLEY U, et al. Meta-analysis of genome-wide scans for human adult stature identifies novel loci and associations with measures of skeletal frame size[J].PLoSGenet, 2009, 5(4): e1000445.
[17] ZHANG L F, ZHOU X, MICHAL J J, et al. Genome wide screening of candidate genes for improving piglet birth weight using high and low estimated breeding value populations[J].IntJBiolSci, 2014, 10(3): 236-244.
[18] WU J H Y, LEMAITRE R N, MANICHAIKUL A, et al. Genome-wide association study identifies novel loci associated with concentrations of four plasma phospholipid fatty acids in the de novo lipogenesis pathway: Results from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium[J].CircCardiovascGenet, 2013, 6(2): 171-183.
[19] OHOKA Y, YOKOTA-NAKATSUMA A, MAEDA N, et al. Retinoic acid and GM-CSF coordinately induce retinal dehydrogenase 2 (RALDH2) expression through cooperation between the RAR/RXR complex and Sp1 in dendritic cells[J].PLoSOne, 2014, 9(5): e96512.
[20] CHENG C X, CUI H Y, ZHANG L, et al. Genomic analyses reveal FAM84B and the NOTCH pathway are associated with the progression of esophageal squamous cell carcinoma[J].GigaScience, 2016, 5: 1.
[21] HERING D M, OLENSKI K, KAMINSKI S. Genome-wide association study for poor sperm motility in Holstein-Friesian bulls[J].AnimReprodSci, 2014, 146(3-4): 89-97.
[22] KIM K T, LEE J S, LEE B W, et al. Association between regulating synaptic membrane exocytosis 2 gene polymorphisms and degenerative lumbar scoliosis[J].BiomedRep, 2013, 1(4): 619-623.
[23] KAESER P S,DENG L B, FAN M M, et al. RIM genes differentially contribute to organizing presynaptic release sites[J].ProcNatlAcadSciUSA, 2012, 109(29): 11830-11835.
[24] RYU J, LEE C. Identification of contemporary selection signatures using composite log likelihood and their associations with marbling score in Korean cattle[J].AnimGenet, 2015, 45(6): 765-770.
猜你喜欢
四川蚕业(2022年2期)2022-11-19
新世纪智能(数学备考)(2021年9期)2021-11-24
中国牛业科学(2020年4期)2020-08-15
世界科学技术-中医药现代化(2020年2期)2020-07-25
四川蚕业(2020年3期)2020-07-16
当代陕西(2019年15期)2019-09-02
猪业科学(2018年5期)2018-07-17
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
文史博览·文史(2015年8期)2015-08-18
