
Epigenetic interventions for brain rejuvenation: anchoring age-related transposons
2018-05-05 06:46AdonisSfera,LisaFayard,CarolinaOsorio等
中国神经再生研究(英文版) 2018年4期
Highlights:
1. Iron and homocysteine accumulation in aging neurons alter genomic methylation.
2. The altered methylome reactivates neuronal cell cycle, enabling transposable element mobilization.
3. miR29/p53 axis restores age-related methylation shifts, reactivating neuronal plasticity.
4. Augmentation of miR-29/p53 axis may preempt neurodegenerative disorders.
Epigenetics refers to the inheritable, non-DNA-related changes in gene expression. Epigenetic mechanisms, including genomic methylation and chromatin condensation, regulate gene transcription and the availability of their products. In general, promoter hypermethylation inhibits, while hypomethylation facilitates adjacent gene expression. With the same token, tight chromatin alignment or heterochromatin inhibits gene transcription, while the looser euchromatin facilitates it.
In contrast to genetic mutations, epigenetic changes are usually reversible,therefore epigenome-modulating therapies may reactivate neuronal plasticity in mature brains (Lennartsson et al., 2015). Furthermore, the recent approval of epigenetic cancer drugs, brought neurodegeneration reversal or delay within reach. Indeed, epi-drugs, including DNA methyltransferase inhibitors (DNMTi), methyl donors and histone deacetylase inhibitors (HDACi) have already entered the clinical practice.
Transposable elements and their anchors:Transposons or transposable elements (TEs) are mobile DNA segments capable of “jumping” from one DNA location to another, increasing the risk of genome destabilization and subsequent neurodegeneration. Indeed, Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD) and frontotemporal dementia(FTD) have been associated with TE mobilization. Moreover, excessive TE“awakening” has been reported in schizophrenia, bipolar disorder, autism and drug addictions. On the other hand, correcting age-related methylome alterations in both neurons and glial cells may lower the specific neurodegenerative disorders triggers.
In addition to their mobilization, lower rates of TE methylation was found in AD. Furthermore, TE activation has been connected to the number and function of mitochondria. Indeed, a novel hypothesis links Alu retrotransposon mobilization in aging neurons with mitochondrial dysfunction and neurodegeneration. This TE self-inserts into the regulatory region of Tom40 gene,altering its product, a mitochondrial protein-trafficking channel (Wylie et al.,2016). As protein import is crucial for mitochondrial survival, its disruption may result in mitochondrial loss, an established early marker of neurodegeneration.
TEs are epigenetically anchored during embryogenesis and remain in“epigenetic restraints” throughout the adult life. DNA methylation, the most studied TE anchoring mechanism, refers to the transfer of methyl groups from of S-adenosyl-methionine (SAM) to the double helix cytosine in a reaction catalyzed by DNA methyltransferases (DNMTs). In this process, SAM is converted to S-adenosylhomocysteine (SAH) and ultimately to homocysteine(HCys). The neurotoxin, HCys, must be rapidly neutralized by remethylation to methionine which in turn provides methyl groups for genomic methylation.HCys accumulation was linked to neurodegeneration and several psychiatric disorders, suggesting a methylome-related pathology in these conditions(Kruman et al., 2004). In addition, HCys accumulation, impairs mitochondrial function as mitochondrial DNA (mtDNA) also requires methylation (Devall et al., 2017).
Old age disrupts the methylome, enabling both TE mobilization and cell cycle reactivation in mature neurons. Indeed, aging was demonstrated to remodel the genomic methylation landscape, imprinting a typical pattern marked by genome-wide hypomethylation and promoter-specific hypermethylation (Mastroeni et al., 2010) (Figure 1). The age-related methylation shift is probably triggered by the intracellular iron and HCys accumulation. Indeed,iron was demonstrated to augment HCys which in return upregulates DNMT 3A/3B and DNMT1, disrupting the methylome.
In contrast, microRNA-29 (miR-29) restores the previous methylation landscape by directly targeting DNMT 3A/3B and DNMT1, while at the same time lowering intracellular iron (Sfera et al., 2017). Interestingly, the selective serotonin reuptake inhibitor (SSRI) paroxetine downregulates DNMT1, suggesting a methylome-protective action. In addition, another SSRI, citalopram was recently shown to lower the progression of mild cognitive impairment (MCI)to AD, likely connecting this drug to methylation repair (Bartels et al., 2018).
Adult neurons, the cell cycle and transposition:Under normal circumstances, neuronal cells do not replicate as they have permanently exited the cell cycle. Pathologically, however genomic damage can induce aberrant replication attempts in mature neurons, an early sign of neurodegeneration. This phenomenon usually results in apoptosis as these cells lack the molecular machinery to adequately complete mitosis. Excess HCys was demonstrated to induce both neuronal cell cycle reentry and TE mobilization, linking these phenomena to genomic methylation (Kruman et al., 2004). This pathology ultimately reflects intraneuronal iron retention as this biometal upregulates HCys. Indeed, HCys plasma level is an established surrogate marker of intracellular iron (Baggott and Tamura, 2015).
Reactivation of the cell cycle in mature neurons is closely connected to transposition and over time to neurodegeneration (Figure 2). Indeed, in somatic cells, TE mobilization only occurs during mitosis, suggesting that replicating postmitotic neurons may be susceptible to transposition (Shi et al.,2007).
Protein 53 (p53) the “genome guardian” blocks both, the neuronal cell cycle reentry and TE mobilization by stabilizing the genome (Wylie et al., 2016).However, to effectively repair the DNA, p53 must be available and active. In the presence of irreparable genomic damage, this pleiotropic protein can trigger apoptosis a phenomenon described in late-stage AD.
Under normal circumstances, p53 is activated by the genomic damage sensors, including the ataxia telangiectasia mutated (ATM) protein. Absent p53 activation, neuronal replication and TE mobilization cannot be averted.Indeed, replicating mature neurons and inactive p53 were documented in ATM knockout rodents. Moreover, neuronal cell cycle reactivation and low ATM protein levels were documented in AD, suggesting that defective ATM methylation may play a role in this disorder (Shen et al., 2016). Aside from being inactive in elderly, p53 is often less available, probably due to promoter hypermethylation and/or SETDB1 histone methyltransferase upregulation, a chromatin-linked enzyme.
MicroRNA-29 reverses age-related methylation changes by directly targeting SETDB1, DNMT 3A/3B and DNMT1 (Figure 2). In addition, miR-29 upregulates p53 and lowers intraneuronal iron and the iron-promoted HCys.Furthermore, miR-29 targets excessive DNA demethylase, methyl groups-removing enzymes, fine-tuning the genomic and mtDNA methylation landscape.Taken together, miR-29 ATM is a genuine “epigenome guardian” which maintains the methylome, preventing age-related alterations. These actions may restore the proper ATM and p53 promoter methylation, preventing neuronal cell cycle reentry and TE mobilization (Figure 2). Indeed, a recombinant premiR-29b was found beneficial in AD, a condition marked by aberrant neuronal replication, excessive transposition and low miR-29 levels (Pereira et al., 2016).Interestingly, the AD-upregulated 12-lipoxygenase (12-LOX), a lipid peroxidation-linked enzyme, was shown to directly suppress miR-29, suggesting that 12-LOX inhibitors could restore the methylome integrity. Therefore, together p53 and miR-29 comprise an anti-aging/anti-neurodegeneration axis which suppresses both aberrant neuronal replication and TE activation, potentially restoring neuronal plasticity in elderly (Figure 2).
Iron chelators as epi-drugs:Iron chelators, including deferasirox, 8-hydroxyquinolones (8HQ) or PBT 434, were found therapeutic in animal models of neurodegenerative disorders. In addition, several natural compounds have iron chelating properties, including curcumin which also augments p53. Moreover,like SSRIs, curcumin downregulates DNMT1, linking this compound to methylation repair (Baggott and Tamura, 2015).
MicroRNA-29 lowers intraneuronal iron by several mechanisms, including ferritin and ferroportin (FPN) stabilization (Sfera et al., 2017). On the other hand, 12-LOX inhibition or lowering its substrate, arachidonic acid (AA) (an omega-6 fatty acid), comprise miR-29 augmentation strategies. Indeed, 8HQ which is both an iron chelator and a 12-LOX inhibitor, has shown benefits in neurodegenerative disorders. For example, Ladostigil, an 8HQ-based drug, was found therapeutic in PD, AD and Lewy body dementia. Furthermore, aside from chelators, iron can be lowered by phlebotomy, thus seniors should be encouraged to routinely donate blood as an iron-lowering intervention. HCys plasma level likely comprises an early marker of neurodegeneration as it accurately reflects the intraneuronal iron.
Lithium, as an epi-drug:Lithium, a drug which has been used in the treatment of bipolar disorder and depression for several decades, has recently demonstrated therapeutic efficacy in neurodegenerative disorders including AD, PD and ALS.
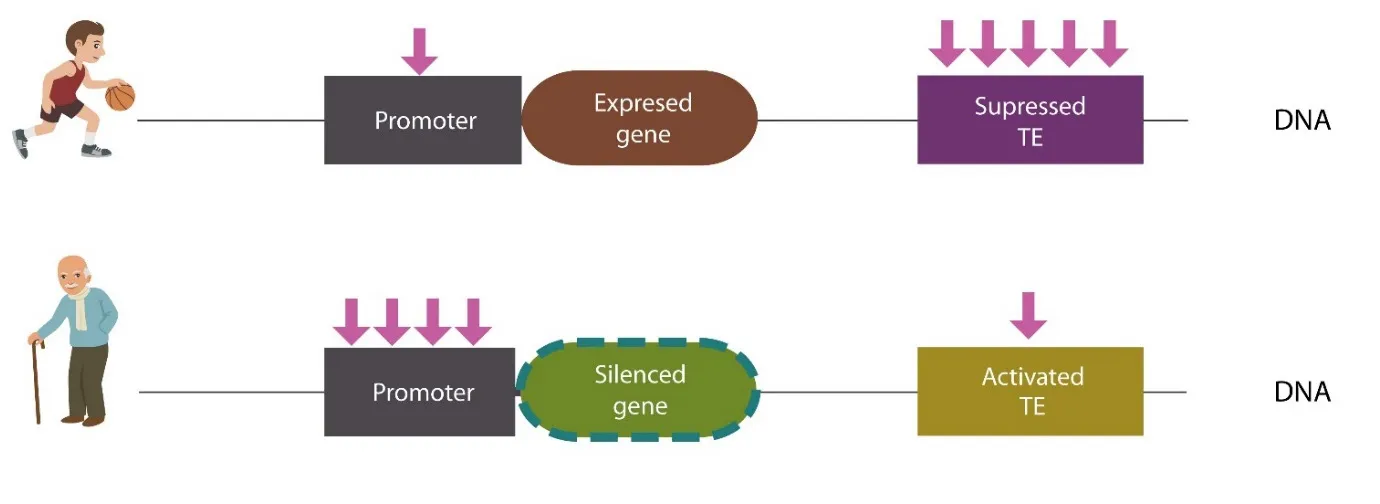
Figure 1 Age-related methylation changes.
From the epigenetic view point, lithium increases global DNA methylation and lowers the methylation of selective promoters, including the brain derived neurotrophic factor (BDNF), enabling its expression. Interestingly,genome-wide hypomethylation and BDNF promoter hypermethylation are the key epigenetic signatures of bipolar disorder. Moreover, lithium inhibits glycogen synthase kinase 3 (GSK3), an enzyme associated with HCys retention and epigenomic damage. Furthermore, lithium facilitates both miR-29b biosynthesis and p53 stabilization, directly augmenting miR-29/p53 axis. These preclinical findings are in line with the recent epidemiologic studies, linking trace lithium in tap water to lower incidence of AD (Fajardo et al., 2018).
Melatonin as an epi-drug: Melatonin is an antioxidant hormone, playing a key role in the regulation of circadian rhythms. At the epigenomic level, melatonin modulates global and promoter methylation. In addition, this hormone upregulates p53, presenting with anti-cancer action. Interestingly, light pollution or excessive night light was demonstrated to destabilize the genome, enabling TE mobilization, probably accounting for the higher incidence of cancer in shiftworkers.
Polyunsaturated fatty acids (PUFAs) as epi-diet:the omega-6/omega-3 ratio:Western diet is characterized by an increase in omega-6 PUFAs and a low intake of omega 3 PUFAs. Impaired omega-6/omega-3 ratio was linked to obesity, neurodegeneration and several psychiatric disorders. The high intake of AA,an omega-6 PUFA, is linked to both iron-induced lipid peroxidation and the upregulation of 12-LOX. On the other hand, omega-3 PUFAs, including eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are neuroprotective,reducing both iron and HCys.
Folate as an epi-vitamin:Folate is crucial for DNA methylation and HCys remethylation to methionine. Despite its presence in numerous foods and supplements, impaired absorption of folate in elderly is common. One common cause of folate deficiency in seniors is alcohol use. Folate depletion leads to increased vulnerability to genomic damage and neurodegeneration. Indeed,epidemiologic studies have associated low folate levels in seniors with MCI and AD. More studies are needed to assess the role of folate supplementation in neurodegeneration prevention, especially as a large epidemiological study linked low maternal folic acid to autism in offspring. In addition, tumor suppression due to cell cycle arrest was linked to high levels of circulating folate,suggesting p53 augmentation.
Conclusions:Epigenetic changes are reversible, opening exciting opportunities for influencing gene expression and neuronal plasticity by manipulating the environment. Epigenomic markers of aging, global DNA hypomethylation and promoter-specific hypermethylation may be engendered by iron and HCys retention. Since we have discussed extensively the role of iron in beta-amyloid toxicity and TE activation in another review, these subjects were not addressed in detail here (Sfera et al., 2017). However, in the long run, neuronal cell cycle activation and TEs mobilization lead to neurodegeneration.
MiR-29/p53 axis may reverse age-related methylomic shifts, stabilizing both the genome and the epigenome, therefore removing a major risk factor of neurodegeneration.
Lowering iron and HCys overload can be accomplishedviachelation, blood donation and maintaining an adequate omega-6/omega-3 ratio.
Adonis Sfera*, Lisa Fayard, Carolina Osorio, Amy Price
Department of Psychiatry, Patton State Hospital, Patton, CA, USA (Sfera A)Department of Psychiatry, Loma Linda University, Loma Linda, CA, USA(Fayard L, Osorio C)
Evidence-Based Medicine, University of Oxford, Oxford, UK (Price A)
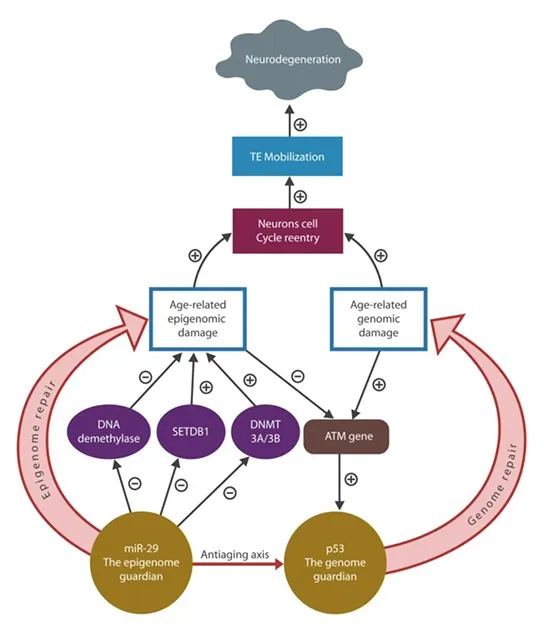
Figure 2 MicroRNA-29 (miR-29) and protein 53 (p53) antiaging axis functions to repair the age-related genomic-epigenomic damage.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer review report:
Reviewer:Ubaldo Armato, University of Verona Medical School, Italy.
Comments to authors:I congratulate the author(s) for the clear brevity of their manuscript adressing such complex topics and trying to unify them into a reasonable picture. The main point I wish to address is that their manuscript covers the alterations which old age may induce into the neuronal methylome. Such alterations might advance neurodegenerative diseases and neuronal death. However, I notice that these same alterations are suggested to be involved in several neurodegerative diseases and not in a single one of them. This implies that the same alterations are likely a part of the old age-linked background which might favor the onset and progression of neurodegenerative diseases yet they are not the specific triggers of diseases as different for instance as AD and Parkinson, etc. Specific triggers for each of such diseases are ignored. Anyway, improving the old-age related aspecific methylome changes should reduce the effectiveness of specific triggers of each neurodegenerative disease and this could have a useful clinical impact (see litium effects, for instance) and entice researchers to address these topics.
Baggott JE, Tamura T (2015) Homocysteine, iron and cardiovascular disease: a hypothesis. Nutrients 7:1108-1118.
Bartels C, Wagner M, Wolfsgruber S, Ehrenreich H, Schneider A, Alzheimer’s Disease Neuroimaging Initiative (2018) Impact of SSRI therapy on risk of conversion from mild cognitive impairment to Alzheimer’s dementia in individuals with previous depression. Am J Psychiatry 175:232-241.
Devall M, Smith RG, Jeffries A, Hannon E, Davies MN, Schalkwyk L, Mill J, Weedon M, Lunnon K(2017) Regional differences in mitochondrial DNA methylation in human post-mortem brain tissue. Clin Epigenetics 9:47.
Fajardo VA, Fajardo VA, LeBlanc PJ, MacPherson REK (2018) Examining the relationship between trace lithium in drinking water and the rising rates of age-adjusted Alzheimer’s disease mortality in Texas. J Alzheimers Dis 61:425-434.
Kruman, II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R, Jr., Gorospe M, Mattson MP (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41:549-561.
Lennartsson A, Arner E, Fagiolini M, Saxena A, Andersson R, Takahashi H, Noro Y, Sng J, Sandelin A, Hensch TK, Carninci P (2015) Remodeling of retrotransposon elements during epigenetic induction of adult visual cortical plasticity by HDAC inhibitors. Epigenetics Chromatin 8:55.
Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J (2010) Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging 31:2025-2037.
Pereira PA, Tomas JF, Queiroz JA, Figueiras AR, Sousa F (2016) Recombinant pre-miR-29b for Alzheimer s disease therapeutics. Sci Rep 6:19946.
Sfera A, Bullock K, Price A, Inderias L, Osorio C (2017) Ferrosenescence: The iron age of neurodegeneration? Mech Ageing Dev doi: 10.1016/j.mad.2017.11.012.
Shen X, Chen J, Li J, Kofler J, Herrup K (2016) Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro doi: 10.1523/ENEURO.0124-15.2016.
Shi X, Seluanov A, Gorbunova V (2007) Cell divisions are required for L1 retrotransposition. Mol Cell Biol 27:1264-1270.
Wylie A, Jones AE, Abrams JM (2016) p53 in the game of transposons. Bioessays 38:1111-1116.

- 中国神经再生研究(英文版)的其它文章
- Acupuncture and neuroregeneration in ischemic stroke
- The adjustment of γ-aminobutyric acidA tonic subunits in Huntington’s disease: from transcription to translation to synaptic levels into the neostriatum
- Bridging the gap: axonal fusion drives rapid functional recovery of the nervous system
- Collagen for brain repair: therapeutic perspectives
- Stimulating effect of thyroid hormones in peripheral nerve regeneration: research history and future direction toward clinical therapy
- Harnessing migraines for neural regeneration