
基于苝二酰亚胺类非富勒烯受体共混体系凝聚态结构调控
2018-05-04 01:42:27韩杰梁秋菊曲轶刘剑刚韩艳春
物理化学学报 2018年4期
韩杰 ,梁秋菊 ,曲轶 ,刘剑刚 ,韩艳春
1长春理工大学,高功率半导体激光国家重点实验室,长春 130022
2中国科学院长春应用化学研究所,高分子物理与化学国家重点实验室,长春 130022
3中国科学院大学,北京 100049
4海南师范大学物理与电子工程学院,海口 571158
1 引言
太阳能电池是将光能转换为电能的装置,而其中有机薄膜太阳能电池(OPVs)由于具有柔韧性好、质量轻、可大面积加工等优势1-7,得到了快速地发展。有机太阳能电池典型的器件结构是由铟锡氧化物(ITO)透明阳极、金属阴极以及夹在他们之间的活性层组成(图 1a所示)。近年来,为提高太阳能电池能量转换效率(PCE),国内外科研工作者在器件结构、活性层材料合成及形貌调控方面做了大量的工作。通过上述优化,目前研究较为成熟的共轭聚合物给体和富勒烯受体组成的体相异质结结构电池的PCE已经突破了11%8,9。

曲轶,1969年生。1991年本科毕业于长春光学精密机械学院应用光学专业;2002年毕业于吉林大学微电子学与固体电子学专业,获博士学位;2003年至2005年在新加坡南洋理工大学材料工程学院做博士后工作。现任长春理工大学高功率半导体激光国家重点实验室副主任,博士生导师。主要从事半导体光电子学方面的研究。


刘剑刚,1982年生。2006年毕业于吉林大学化学专业;2011年毕业于中国科学院长春应用化学研究所高分子化学与物理专业,获得博士学位。现任中国科学院长春应用化学研究所副研究员,主要从事聚合物太阳能电池活性层凝聚态结构调控方面的研究。 韩艳春,1966年生。1990年本科毕业于中国科学技术大学高分子物理专业;1995年毕业于中国科学院长春应用化学研究所高分子化学与物理专业,获博士学位;1996年至1998年在德国凯泽斯劳腾大学为德国洪堡基金会资助的Research Fellow;1999年至2000年在美国密执安大学做博士后工作。现任中国科学院长春应用化学研究所研究员,博士生导师。主要从事高分子薄膜表面形态结构和功能构效关系方面的研究。
根据受体材料的不同,有机太阳能电池主要分为聚合物/富勒烯体异质结太阳能电池、聚合物/聚合物体异质结太阳能电池及聚合物/非富勒烯体异质结太阳能电池。然而,由于富勒烯分子在可见光范围内光子吸收效率低,限制了光伏电池的短路电流(Jsc);另外,富勒烯分子的最低未占据分子轨道(LUMO)能量较低,与聚合物共混后能量损失较大,从而开路电压(Voc)较低。上述两点因素限制了聚合物/富勒烯体系太阳能电池性能的进一步提高10,11。对于聚合物/聚合物共混体系而言,聚合物给受体分子间缠结严重及分子间作用力较大等因素,不利于相区纯度及相区尺寸的调节。而非富勒烯小分子受体则能够很好的克服上述两个体系的不足:首先,可以通过对非富勒烯受体分子结构进行灵活的设计,增加共混薄膜在可见光和近红外光光谱范围内的光吸收强度,可显著提高器件的 Jsc;其次,通过调节受体材料的LUMO,提高器件的Voc;另外,非富勒烯小分子受体与聚合物给体分子间作用力弱,且分子易于迁移扩散,形貌调控简便易行。正是由于聚合物/非富勒烯体系太阳能电池具有众多优点,目前,非富勒烯受体受到科学家们的广泛关注12-16。1985年,Tang17采用苝四酰二亚胺为电子受体材料首次制备了非富勒烯有机太阳能电池,其效率仅为0.95%。近年来,随着分子结构及形貌调控的不断优化,聚合物/非富勒烯体系太阳能电池已经有了显著提高。2017年,Hou课题组16,18将新的聚合物给体材料(PBDB-T-SF)与新的小分子受体(IT-4F)共混,制备的器件PCE已经达到13.1%;进一步佐证了聚合物/非富勒烯太阳能电池具有光明应用前景!

图1 (a)有机太阳能电池结构示意图;(b)有机太阳能电池工作原理图Fig.1 (a) The structure of organic solar cells; (b) the scheme of organic solar cells work processes.
聚合物/非富勒烯太阳能电池中,给受体均具有较强的光吸收能力,两组分均能形成激子,进而为光电转化过程做出贡献,如图1b所示。其能量转换过程主要包括以下几个步骤:光子吸收、激子扩散、电荷转移态分离、电荷传输以及电荷收集五个过程19。首先,光子被活性层吸收后,给体材料和受体材料上的电子会被激发,与空穴形成激子。紧接着激子在浓度梯度作用下扩散迁移至给受体界面处形成电荷转移态(CT态)。CT态是被库仑引力束缚的电子-空穴对,在界面处电势的驱动下给体产生的 CT态中的电子将从给体LUMO能级向受体的 LUMO能级转移(Channel-I);而受体产生的CT态中的空穴将从受体最高占据分子轨道(HOMO)能级向给体的HOMO能级转移 (Channel-II)。解离后的自由电荷在内建电场的作用下分别沿着给体/受体材料形成的连续电荷传输通道传输到阳极/阴极,从而完成电荷传输过程20。从有机光伏电池的工作原理,我们不难发现,给体/受体材料形成的活性层薄膜形态对光伏电池的最终性能具有至关重要的影响21-23。活性层相分离结构决定了载流子收集效率及双分子复合程度:当给受体均形成连续通路时,载流子收集效率高,可提高Jsc;相区尺寸决定激子扩散效率及载流子复合程度,当相区尺寸过大时会导致激子扩散效率低,不利于提高Jsc。相区纯度决定激子分离及载流子传输:当相区纯度低时,载流子传输过程中复合较为严重,不利于提高填充因子(FF);当相区纯度高时,给受体界面处CT态分离困难,不利于提高Jsc。
根据分子结构特点,较常见的非富勒烯小分子受体材料大体分为五大类。其中包含苯噻唑类小分子(Vinazene)、多环芳烃荧蒽结合酰亚胺结构为主体的衍生物(Fluoranthene-fused imide (FFI) based)、芴基衍生物(Fluorene and dithienylsilole based)、并吡咯二酮类小分子(Diketopyrrolopyrrole (DPP) based)及萘二酰亚胺(Naphthalene diimide (NDI))/苝二酰亚胺(Perylene diimide (PDI))类小分子24-31。PDI类小分子是目前种类最广也是最为成功的一类非富勒烯受体材料。一方面酰亚胺类材料具有良好的光、热和气候稳定性、抗腐蚀性、化学惰性、光吸收特性以及较高的荧光量子效率等特点,另一方面该类材料具有大共轭平面结构以及两个亚胺环结构,因而表现出高的电子亲和势和很强的吸电子能力。研究工作者不断优化材料结构,PDI类的材料由原本单一的刚性稠环结构(如 N,N’-bis(1-ethylpropyl) perylene-3,4,9,10-tetracarboxylic diimide, 简 称EP-PDI),逐渐演变为具有扭曲结构/二维结构的PDI材料(如 twisted PDI以及 Bis-PDI-EG 等),伴随着分子堆积的变化,相应的电荷传输性质也显著改善32,33。目前 PDI类的非富勒烯体系通过分子结构、薄膜形态以及界面层的优化,光伏效率得到大幅提高,许多体系的光伏效率都达到了6.0%以上34,35。
然而对于传统的 PDI小分子材料而言,共混薄膜的相分离形貌远未达到理想光伏器件的形态要求:一维 PDI分子难于形成连续通路,严重限制了电子的传输与收集过程;PDI与给体材料相容性差异大,相区尺寸难于调控,不利于激子分离或载流子传输;PDI为受体时,共混体系形成的CT态间束缚能高,需要控制共混相含量,降低单分子及双分子复合几率。针对以上问题,我们从分子间相互作用的角度入手,通过调节给受体比例及分子扩散速率实现互穿网络结构构筑。在此基础上,通过调节给受体分子间相互作用及溶质分子与溶剂分子间相互作用,实现了高相容性及低相容性共混体系相区尺寸的可控调节。调节溶剂与PDI分子间相互作用,控制PDI分子在溶液中固-液相分离程度,实现高相容性及低相容性共混体系共混相含量的可控调节。
2 调节给受体比例及分子扩散速率构建互穿网络结构
光伏电池中给受体形成双连续通路可确保电荷转移态分离及自由载流子传输,而孤岛状相分离的存在则增加了载流子双分子复合几率,增加空间限制电荷密度。Chiu等通过经典体系聚(3-己基噻吩)(P3HT)/富勒烯衍生物(PCBM)共混体系,系统的研究了双连续结构对载流子传输的影响36。结果表明,当富勒烯含量较低时(例如质量百分比为38%),P3HT可形成连续的空穴通路(空穴迁移率大于 1.0 × 10-4cm2·V-1·s-1),而富勒烯无法形成连续通路(电子迁移率低于 1.0 × 10-5cm2·V-1·s-1),此时器件PCE仅为3.06%;当其含量升高至44%时,富勒烯形成连续电子通路,此时电子迁移率急剧升高至 5.0 × 10-4cm2·V-1·s-1,器件 PCE 也达到最大值3.70%;当富勒烯含量继续升高(55%),此时过量的富勒烯聚集会破坏 P3HT形成的空穴通路,导致器件PCE下降至2.80%。在共轭聚合物共混体系中,Sepe等37,38对相分离结构做了初步研究,结果同样表明只有当给受体均形成连续相,才有利于器件性能的优化。由此可见,构筑给体/受体共混体系双连续相分离结构,为空穴及电子提供传输的连续通路,确保其有效传输至相应电极,是制备高效光伏电池的重要前提。
与富勒烯受体共混体系类似,在非富勒烯受体共混体系中给受体比例及其自组织能力均直接影响互穿网络结构的形成。Meredith课题组39利用差示扫描量热法(DSC)通过监测 P3HT/2- [(7-{9,9-di-n-propyl-9H-fluoren-2-yl}benzo[c][1,2,5]thiadiazol-4-yl)methylene]malononitrile (K12)共混体系给体材料和受体材料熔融温度的变化绘制出了两元体系相图,并且建立了给受体比例-共混相分离结构-器件性能间的关系。结果表明,P3HT/K12体系为简单的低共熔体系,低共熔点时P3HT的质量分数(Ce)约为75%。当P3HT浓度为C = Ce时,P3HT含量高,将抑制K12聚集结晶,从而无法形成连续的电子通路。而当 P3HT的质量分数降至33%时,此时K12过量,易于成核进而发生结晶,形成互穿网络结构;此时,电子与空穴能够顺利传输至相应电极,器件 Jsc及 PCE均达到最高值。对于共混程度较高的 PDI体系,PDI结晶受到给体分子抑制,限制共混体系形成互穿网络结构。例如,7,7’-(4,4-bis(2- ethylhexyl)- 4H-silolo[3,2-b:4,5-b’]dithiophene-2,6-diyl)bis(6-fl uoro-4-(5’-hexyl-[2,2’-bithiophen]-5-yl)benzo[c][1,2,5]thiadiazole) (p-DTS(FBTTh2)2)与 EP-PDI 是典型的具有良好相容性的非富勒烯体系。在该体系中,分散在给体中的EP-PDI受体分子无法构筑有效的电子传输通道,因而只能扮演电荷捕获陷阱的角色。因此,目前大量工作集中于利用添加剂(SAs)40-42、热退火(TA)43,44及溶剂退火(SVA)33,45等手段促进 PDI聚集结晶,从而构建互穿网络结构。
尽管人们意识到了给受体比例及受体自组织能力对互穿网络的形成有影响,但是对不同比例下形貌形成起因及互穿网络形成机理的认识尚不清晰。本部分工作中以 p-DTS(FBTTh2)2/EP-PDI共混体系为研究对象,指出成膜过程中的液-固相分离是不同给受体比例下获得不同形貌的主要原因;同时指出通过TA温度控制分子扩散速率,利用结晶限制相分离的方法可获得纳米级互穿网络结构46。
如图 2所示,当 p-DTS(FBTTh2)2含量超过80%或EP-PDI含量超过60%时,p-DTS(FBTTh2)2或EP-PDI自组织形成大尺寸聚集;而给受体比例在 7 : 3 到 5 : 5 之间时,旋涂薄膜几乎无明显相分离结构。众所周知,p-DTS(FBTTh2)2和 EP-PDI均为平面性分子,结晶能力较强。当给受体含量差异较大时,例如给体含量为80%,由于EP-PDI分子对 p-DTS(FBTTh2)2的干扰较小,p-DTS(FBTTh2)2在成膜过程中易聚集成核从而形成液-固相分离;随着溶剂挥发,液-固相分离程度增加,p-DTS(FBTTh2)2生长成为大尺寸晶体,破坏了互穿网络结构形成。与此类似,当EP-PDI含量较高时,EP-PDI发生固-液相分离,同样也会抑制互穿网络结构的形成。而当给受体比例相接近时,以 p-DTS(FBTTh2)2/EP-PDI为 6 : 4 为例,由于给受体分子间相容性较高相互抑制结晶,从而形成结晶度较低的无定型薄膜。我们利用TA处理方法在不同温度下分别对上述无定型薄膜进行TA处理:当T < 90 °C时,薄膜形貌基本保持不变,无互穿网络结构形成;当 90 °C < T < 130 °C时,p-DTS(FBTTh2)2分子自组织形成结晶网络骨架,EP-PDI分子形成微晶,最终形成纳米级互穿网络结构;当 T > 130 °C 时,EP-PDI形成大尺寸晶体,突破p-DTS(FBTTh2)2结晶网络骨架,从而破坏了互穿网络结构。

图2 不同比例和不同温度下热退火10 min的p-DTS(FBTTh2)2/EP-PDI共混薄膜相图46Fig.2 Phase diagram of p-DTS(FBTTh2)2/EP-PDI blend films at different ratios and TA treated at different temperatures for 10 min46.
我们利用荧光光谱通过分子扩散速率分析说明形貌对 TA处理温度的依赖性,如图 3所示。p-DTS(FBTTh2)2/EP-PDI共混薄膜荧光猝灭非常严重,薄膜几乎无相分离结构。TA温度T升高,EP-PDI及p-DTS(FBTTh2)2荧光信号增强,表明薄膜相分离程度增加。其中,610 nm附近的荧光峰对应于EP-PDI分子聚集,730 nm附近的荧光峰对应于p-DTS(FBTTh2)2的聚集,我们分别研究了EP-PDI及p-DTS(FBTTh2)2荧光强度与TA处理温度的依赖关系,如图3b所示。两者变化曲线按照斜率均可以分为两个区间:对于EP-PDI的分界点为 90 °C,而对于 p-DTS(FBTTh2)2的分界点为130 °C。根据 Arrhenius公式(1):

D为扩散系数,Do是温度无关的频率因子,R是通用气体常数,T是开尔文绝对温度,Ea是活化能。分子的扩散速率具有温度依赖性,当TA温度T < 90 °C 时,EP-PDI与 p-DTS(FBTTh2)2分子扩散能力均较弱,因此无法聚集结晶促进互穿网络形成。当 90 °C < T < 130 °C 时,p-DTS(FBTTh2)2分子扩散能力进一步增加,促进发生结晶形成结晶网络结构;与此同时,EP-PDI分子进入熔融态(其熔融温度为73 °C)扩散能力急剧升高;然而,由于 p-DTS(FBTTh2)2晶体网络的限制,EP-PDI仅能在网络间形成小尺寸晶体,从而最终形成纳米级互穿网络结构。当温度进一步升高T > 130 °C时,由于温度高于共混体系中p-DTS(FBTTh2)2晶体的熔融温度,p-DTS(FBTTh2)2分子的扩散速率也大幅增加,导致p-DTS(FBTTh2)2晶体网络被破坏,无法限制EP-PDI分子聚集,从而形成大尺寸晶体。

图3 (a)不同温度下热退火10 min的p-DTS(FBTTh2)2/EP-PDI(6 : 4)共混薄膜荧光光谱图;(b)不同温度下热退火的p-DTS(FBTTh2)2/EP-PDI(6 : 4)共混薄膜620和725 nm处的荧光强度变化图46Fig.3 (a) Fluorescence spectra of p-DTS(FBTTh2)2/EP-PDI blend films at 6 : 4 TA at different temperature for 10 min;(b) evolution of fluorescence intensity at 620 and 725 nm of p-DTS(FBTTh2)2/EP-PDI blend films at 6 : 4 TA treated at different temperature46.
当 p-DTS(FBTTh2)2含量超过 80%或 EP-PDI含量超过60%时,固-液相分离将诱导大尺寸相分离发生,不利于激子分离;给受体比例介于7 : 3到5 : 5之间,共混薄膜无明显相分离结构,载流子无法传输至相应电极。而通过TA处理控制分子扩散速率可获得 p-DTS(FBTTh2)2/EP-PDI共混体系纳米级互穿网络结构,此时即能保证激子扩散至界面分离,又能促进载流子传输至相应电极,器件PCE及各项参数如表1所示。
3 调节相区尺寸平衡激子分离与载流子传输
相区尺寸直接决定载流子成对复合及非成对复合几率。众所周知,激子为被库仑引力束缚的电子-空穴对,其寿命一般为 100 ps-1 ns,如果激子不能在寿命期限内扩散至给受体界面,则只能衰减回到基态发生成对复合。由此可见,相区尺寸应尽量减小,从而保证激子均能扩散至界面。另外,载流子要扩散至相应的电极除要求给受体形成连续通路外,给受体相区尺寸不能过小,因为当相区尺寸小到低于库仑捕获半径时,电子与空穴则难以摆脱库仑引力,最终导致成对复合的发生。因此,对于相容性差的PDI共混体系而言,
应当降低相分离程度,而对于相容性好的 PDI共混体系则应当增加相分离程度。本部分工作中,我们通过调节给受体间相互作用,实现了相分离程度的可控调节。在给受体相容性差的体系,通过增加溶剂溶解度,增加给受体间相互作用,降低相区尺寸;在给受体相容性好的体系,在促进分子结晶的基础上,引入给受体间电荷转移作用,抑制PDI过分聚集,从而形成纳米级互穿网络结构。
3.1 增加给受体间相互作用减小相分离程度
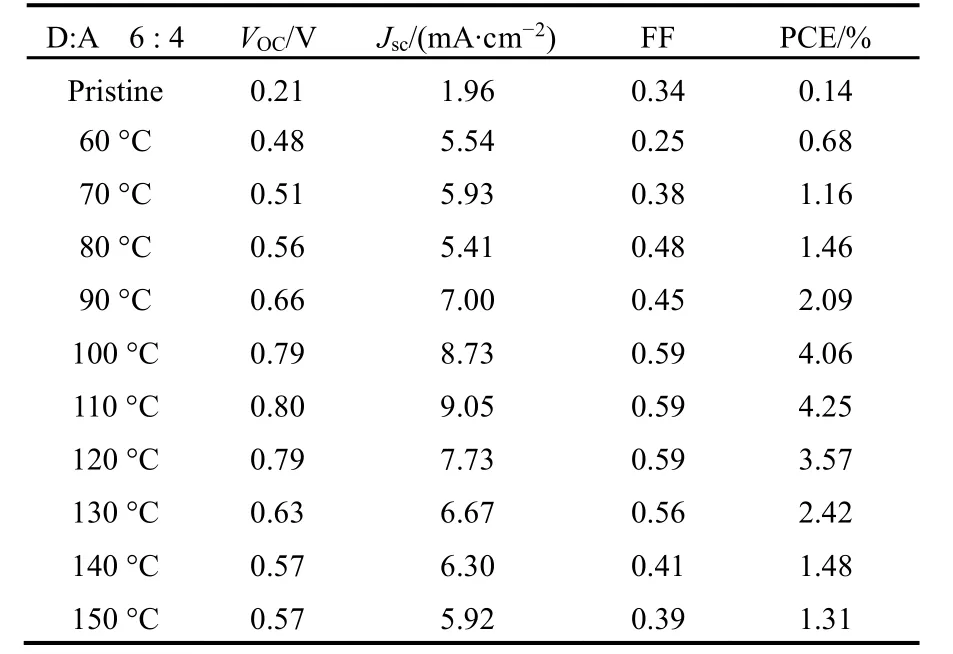
表1 不同温度热退火10 min的p-DTS(FBTTh2)2/EP-PDI(6 : 4)器件参数46Table 1 Device parameters of p-DTS(FBTTh2)2/EP-PDI of 6 : 4 ratio subjected to TA at different temperatures for10 min46.
对于相容性差的 PDI共混体系而言,应当降低相分离程度。正如前面提到的,具有平面结构的PDI小分子由于具有更强的π-π作用,非常容易形成大尺寸的聚集体。降低 PDI分子间的共平面堆积作用可有效地降低 PDI的聚集尺寸并实现器件性能的改善32,47。Narayan课题组32以联氨单键连接两个 PDI单元合成了具有扭曲结构的 PDI分子,有效降低了 alkylthiophene-2-yl-substituted benzo[1,2-b:4,5-b’]dithiophene (PBDTTT-CT)/PDI共混体系的相分离程度。此外,人们还尝试从PDI分子内核的1,6,7,12取代位构建连接基团,从而形成更多具有扭曲结构的PDI二聚体33,35,41,48-50、三聚体51甚至四聚体52,53,这些新颖的分子结构不仅有效抑制了 PDI分子聚集,而且拓宽了自由电子的传输通道(由一维转变为二维甚至三维),因此大大提高了载流子迁移率,光伏效率也得到了显著提升。目前文献报道的器件性能最高的 PDI非富勒烯小分子体系就是 Wang课题组54基于上述思想构建的结构扭曲的PDI二聚体,其光电转化效率已经达到了 7.16%。此外,人们也尝试通过各种物理手段来限制非富勒烯小分子体系的相分离程度、增加两组分的相容性。Fréchet课题组55向P3HT/PDI体系中引入P3HT与PDI的嵌段聚合物作为相容剂。为了减小界面张力,该嵌段聚合物倾向于分布在两相界面处,并且与给体、受体分子都存在相互作用。相容剂的引入有效降低了PDI组分的相区尺寸,同时共混薄膜相分离形貌的稳定性也得到了提高。
在P3HT/EP-PDI体系中,EP-PDI平面型结构导致强的 π-π相互作用,容易形成微米级的聚集体。共混体系的相分离过程及薄膜形貌是由材料性质和溶剂性质共同决定,为此我们采用溶剂SAs氯萘(CN)优化薄膜形貌56。如图4所示,由于P3HT与EP-PDI相容性差,二者皆趋向于自组织形成大尺寸的相分离形貌。当加入少量CN时(< 1.0%),可抑制P3HT及EP-PDI大尺寸聚集。而当引入超过1.0%的CN时,P3HT及EP-PDI的聚集尺寸未继续降低,反而出现了更大尺寸的相分离。为了进一步研究CN添加剂对活性层的结晶性的影响,我们对P3HT/EP-PDI共混薄膜进行了掠入射X射线衍射(GIXD)的表征。在GIXD谱图上,EP-PDI分子的(200)和(400)晶面与 P3HT 的(100)和(200)晶面对应的衍射峰位非常接近57。因此,在对共混薄膜的结晶性进行表征之前,我们采用丙酮溶剂浸泡的方法首先将 EP-PDI组分洗去。结果表明,体系中引入体积分数为0.5%或者0.75%的CN时,对 P3HT的有序堆积及结晶性产生了一定程度的负面影响。当加入0.75%的CN时,P3HT的结晶强度以及结晶尺寸都降到最低58-60,当继续增加CN含量,P3HT的结晶强度以及结晶尺寸随之增加。如图5b中,我们总结归纳了P3HT的(100)衍射峰强和晶体尺寸(La)随着CN添加剂浓度的变化情况。

图4 CB中加入不同含量CN添加剂的P3HT/EP-PDI (1 : 1)共混薄膜透射电镜图和原子力显微镜图56Fig.4 TEM and AFM images of P3HT/EP-PDI (1 : 1) blend films prepared by using different concentrations of the solvent additive CN in CB solutions56.(a, a′) without CN; (b, b′) 0.5% CN; (c, c′) 0.75% CN; (d, d′) 1.0% CN; (e, e′) 1.25% CN; (f, f′) 1.5% CN. The insets of TEM images show the corresponding SAED patterns. The bottom panels show the typical cross-sectional height profiles of P3HT/EP-PDI blend films prepared under different conditions.

图5 (a) CB中加入不同含量CN的P3HT/EP-PDI (1 : 1)共混薄膜掠入射X射线衍射图(经过丙酮处理);(b)对应P3HT(100)取向的衍射峰强和晶体尺寸图56Fig.5 (a) GIXD patterns of the P3HT/EP-PDI (1:1) blend films after acetone-soaking treatment. The films were prepared by using different concentrations of the CN additive in CB solutions. (b) Plots of the P3HT diffraction peak intensity and the crystallite size (La) corresponding to the (100) plane versus the concentration of CN56.The inset illustrates the lamellar supramolecular structures formed by P3HT chains.
考虑到P3HT和EP-PDI在CN溶剂中都表现出良好的溶解性,我们推测 CN添加剂的加入可以有效促进EP-PDI分子向P3HT相的扩散,从而抑制了 P3HT的结晶过程。由于这种两相分子间的扩散作用使EP-PDI的聚集作用减弱,因此相应的相区尺寸也得到了明显的降低。而当引入超过1.0%的 CN时,活性层的成膜时间相应变长,此时P3HT以及EP-PDI的自组织过程变得显著,共混体系相分离的驱动力逐渐增大,最终导致了更大尺寸的相分离现象的发生。由于在该共混体系中,给体(P3HT)与受体材料(EP-PDI)都具有较强的结晶能力,因此可想而知,这种相分离发生的驱动力必然远远高于富勒烯体系。或许这就是基于PDI的非富勒烯体系中的最优SAs浓度(0.75%)远低于富勒烯体系常见的最佳条件(2%-3%)的原因40,61-63。基于此,我们不难发现,非富勒烯小分子共混体系需要更为严苛的调控条件才能达到理想的相分离形态。如表2所示,加入0.75%的CN使器件的 Jsc提高了 5倍(由 0.26 mA·cm-2提高到1.45 mA·cm-2)。器件性能的提高主要归因于 CN添加剂明显优化了活性层的相分离尺寸,从而使激子分离效率得到改善。EP-PDI相区尺寸的降低,不仅促进了电荷转移过程而且也抑制了稳定的分子间态的形成,最终起到了改善电荷分离过程的作用64。
3.2 调节给受体间相互作用增加相分离程度
对于共混程度较高的非富勒烯小分子共混体系,人们利用各种常见的后处理手段调控共混薄膜的有序聚集程度,从而实现自由电荷传输路径的优化。例如,在基于PDI二聚体的非富勒烯体系中,Yao课题组33利用混合溶剂蒸汽处理的方法显著提高的给体分子以及受体分子的自组装行为并优化了薄膜的相分离程度,最终器件的 PCE由1.44%提高到了6.08%。此外,Keivanidis课题组44还采用TA的手段调控PDI体系受体相的聚集程度,并利用Raman光谱结合密度泛函理论表征了不同TA条件下PDI的π-π堆积强度的差异。人们发现,不同TA温度下的Raman峰强比与荧光光谱表现出一致的变化规律,即 TA能够增强PDI分子间 π-π堆积作用,从而促进受体组分形成更多有序聚集相、提高电子迁移率。另外,通过控制分子在溶液中的聚集程度是调节相分离程度的有效手段。例如以共轭聚合物给体(PIDT-DTN)/小分子受体(ITIC)共混体系为活性层制备的器件性能仅为 1.1%,而以共轭聚合物(PBDT-DTN)/ITIC共混体系为活性层的器件性能高达 8.3%。这是由于 ITIC分子有序堆叠能力较弱,因此当与聚集能力较差的 PIDT-DTN分子(indacenodithiophene (IDT)单元的烷基侧链对于链间聚集具有很大的位阻作用)共混后,体系相分离驱动力较低,无法获得纳米互穿网络结构。而ITIC与聚集能力较强的PBDT-DTN分子共混后,由于PBDT-DTN分子间有序堆叠能力强,在溶液中可形成晶核,利于成膜过程中进一步结晶,从而促进相分离结构的形成65,66。然而,上述增强薄膜相分离程度的方法中,在促进给体聚集的同时,由于PDI分子间强π-π相互作用,其倾向于形成大尺寸聚集;在大尺寸的 PDI聚集体中往往容易形成稳定的分子间态,进一步降低了激子分离效率64。因此,如何促进给受体聚集的同时,抑制 PDI形成大尺寸聚集,是进一步提高器件性能的关键。

表2 不同含量CN添加剂制备的P3HT/EP-PDI (1 : 1)混合太阳能电池光伏特性56Table 2 Photovoltaic properties of P3HT/EP-PDI (1 : 1)blend solar cells fabricated with different concentrations of the CN additive56.
在我们的工作中,利用良溶剂蒸汽处理手段,通过向共混体系引入与 D-A分子间电荷转移作用,抑制PDI聚集,得到了PDI聚集程度适中的互穿网络结构,改善了激子分离与电荷传输效率,最终的器件性能由不足0.2%提高到超过3.0%67。SVA或 TA等是改善活性层形貌的主要手段。考虑到各种溶剂氛围对分子间相互作用程度的差异,我们选择了氯苯(CB)、邻二氯苯(ODCB)与乙酸乙酯(EA)三种溶剂进行SVA处理对比研究。这三种溶剂的SVA处理在接下来的描述中将分别被简写为CB-SVA、ODCB-SVA以及EA-SVA。其中,CB与ODCB既是p-DTS(FBTTh2)2的良溶剂,同时也是EP-PDI的良溶剂。而EA只对EP-PDI具有一定的溶解性,对p-DTS(FBTTh2)2则是劣溶剂。我们采用透射电子显微镜(TEM)以及原子力显微镜(AFM)照片对退火前后的 p-DTS (FBTTh2)2/ EP-PDI共混薄膜的相分离形貌进行表征,如图6所示。与薄膜形态平整的初始薄膜(薄膜粗糙度小于1 nm)相比,退火处理后的共混薄膜粗糙度显著增强。其中,变化最显著的为EA-SVA以及TA处理后的共混薄膜,其粗糙度提高到了10 nm以上。此外,选区电子衍射(SAED)谱图中衍射环的出现表明,所有的后处理手段都有效提高了薄膜结晶性。尽管各种处理手段都能够促进结晶纯相区的形成,但是TA处理后产生了数百纳米的相分离,而如此大尺寸的相分离并没有出现在SVA处理的共混薄膜中。
利用面内 X射线衍射(XRD)我们进一步表征了退火处理后 p-DTS(FBTTh2)2与 EP-PDI的 π-π堆积作用随退火时间的变化,如图 7所示。我们发现,随着退火时间的延长,对应两组分的 π-π堆积强度逐渐增强。为了获得退火处理对薄膜结晶性的定量信息,我们利用Scherrer公式58计算得到了两组分沿着面内π-π堆积方向上的 La,如图7(c, d)所示。通过分子间π-π方向所对应的La随退火时间的变化规律,我们可以近似地总结出不同处理条件下的结晶动力学的差异。我们发现,随着 SVA或者 TA处理时间的延长,尽管p-DTS(FBTTh2)2的La都表现出逐渐增大的变化趋势,但是不同处理手段下的La却存在较大差异。从图可以看出,在 CB-SVA处理过程中,两组分的分子间π-π堆积方向的La始终保持在7-10 nm之间。而在TA处理过程中,共混薄膜中的两组分的La基本处于10-13 nm之间。这一结果表明,选择性溶剂氛围(CB或者 ODCB)不仅保障了p-DTS(FBTTh2)2/EP-PDI共混薄膜中形成足够的结晶纯相区,而且还能够促进D-A分子接触、增强D-A作用,从而起到限制La以及调控相分离形态的作用。而在 TA以及 EA-SVA处理条件下,p-DTS(FBTTh2)2与 EP-PDI两组分的结晶过程相对独立地完成,因此共混薄膜往往形成更大尺寸的晶体以及大的相分离程度。
如表 3所示,未处理的原始器件表现出非常低的光伏性能,Voc为 0.68 V,Jsc为 0.39 mA·cm-2,FF为0.40,最终的PCE值只有0.11%。我们将较低的Jsc值归因于有效电荷传输通道缺失以及结构无序的薄膜形态。通过 TA处理后,Jsc值显著提高到了5.23 mA·cm-2,与此同时最终的PCE值也达到了 1.82%。通过 CB-SVA处理后的光伏器件表现出更为优异的器件性能,Voc为 0.76 V,Jsc为 6.75 mA·cm-2,FF 为 0.59,最终的 PCE 值超过了 3.0%。正如我们之前提到的,CB-SVA与ODCB-SVA对两组分的自组装过程表现出类似的效应。该类溶剂氛围诱导下的相分离形态不仅提供了必要的电荷传输通道,而且保证了给体/受体材料必需的接触界面,因此取得了最优的器件性能。而在EA-SVA条件下,与TA处理效果类似,给体/受体自组装形成固体有序结构的过程相对独立地进行,共混薄膜表现出较大的相分离形貌,激子的分离效率因此大打折扣,最终的器件性能只是因为电荷传输效率的优化而得到一定程度的改善。

图 6 p-DTS(FBTTh2)2/EP-PDI (1 : 1)经(a, a′)沉积,(b, b′)CB 蒸汽处理、(c, c′)ODCB 蒸汽处理、(d, d′)EA 蒸汽处理和(e, e′)TA处理的共混薄膜透射电镜图和原子力显微镜高度图以及透射电镜对应的选区电子衍射图67 Fig.6 TEM and AFM (5 mm × 5 mm) height images of (a, a′) as deposited, (b, b′) CB-SVA treated, (c, c′) ODCB-SVA treated, (d, d′) EA-SVA treated and (e, e′) TA treated p-DTS(FBTTh2)2/EP-PDI (1 : 1) blend films. The insets of TEM images show the corresponding SAED patterns67.

图7 经后处理的p-DTS(FBTTh2)2/EP-PDI (1 : 1)共混薄膜非原位面内X射线衍射图和相应的 π-π相互作用分子晶体尺寸图67 Fig.7 Ex situ in-plane XRD of p-DTS(FBTTh2)2/EP-PDI (1 : 1) blend films after post-treatments. Plots of intermolecular π-π crystallite size (La) in blend films as a function of annealing time in the direction of in-plane67. (a and c) CB-SVA post-treatment, (b and d) TA post-treatment. The peaks corresponding to the π-π stacking of p-DTS(FBTTh2)2 and EP-PDI were used for calculation, respectively.

表3 不同后处理方式加工的p-DTS(FBTTh2)2/EP-PDI(1 : 1)混合太阳能电池光伏特性67Table 3 Photovoltaic properties of p-DTS(FBTTh2)2/EP-PDI (1 : 1) solar cells processed under different post- treatment methods67.
4 调节共混相含量增加CT态分离效率
除了给受体形成互穿网络结构及适宜的相区尺寸外,给受体共混后形成的共混相对器件性能的影响也至关重要68-71。McGehee课题组发现68,69,72,P3HT/PCBM共混薄膜是由给体纯相、受体纯相以及给体/受体共混相组成。这样的三相模型所构建起的瀑布式的能级结构大大增加了界面处的电荷分离效率。与富勒烯体系较高的内量子效率(75%-90%)相比,P3HT(或聚双己基噻吩(PDHTT))与苯噻唑类小分子 HPI-BT组成的非富勒烯共混薄膜的内量子效率却只有56%73。共混相所产生的能级相比于纯相会发生位移,因此在共混相与纯相之间会产生能级梯度。该能级梯度有助于光致产生的电子和空穴对的空间分离71,74。因此,在保证给受体有序聚集形成互穿网络结构的基础上引入共混相,构筑给体纯相、受体纯相及给受体共混相三相模型,是进一步提高器件性能的关键。
对非富勒烯体系而言,薄膜中两组份的相容性以及聚集行为的调控可以通过分子设计与加工方法相结合的形式实现75,76。例如,一种窄带隙的p型聚合物通过改变共轭侧链结构,改善了与NDI类n型聚合物的相容性,从而增加了共混相含量;在此基础上继续引入 1,8-二碘辛烷后,可促进 n型聚合物结晶,从而构筑了三相模型形貌,最终实现了器件性能的显著改善76。与通过复杂的设计合成调控分子相容性相比,引入特定溶剂 SAs实现共轭分子间的相互作用的改变更为简单有效,而且同样可以实现共混相与结晶纯相比例的调控。尽管SAs实现薄膜形态调控的手段已经被广泛使用,而且人们在特定富勒烯体系中较为深入地研究了SAs的选择标准和使用条件77-82,然而对于非富勒烯体系而言,对SAs的选择标准仍然缺少系统的研究,SAs的性质与薄膜微结构的调控之间具有怎样的关系仍不清楚。
在本部分工作中,我们以EP-PDI非富勒烯体系作为研究对象,采用选择性SAs手段分别实现了高相容性及低相容性活性层的三相组成的调控。对共混程度较低的PTB7/EP-PDI体系和具有良好相容性的 p-DTS(FBTTh2)2/EP-PDI体系,分别加入 EP-PDI的良溶剂和劣溶剂,利用选择性SAs获得了组成更为平衡的共混相和结晶纯相,从而改善了激子分离以及电荷传输。最终,我们通过对比研究提出了基于 PDI的非富勒烯体系的SAs选择标准,并研究了特定SAs在优化三相(无定型共混相、给体结晶纯相和受体结晶纯相)体异质结形貌过程中所起到的作用。
我们采用溶度参数(δ)来评估溶剂SAs与活性层材料之间的相容性;采用EP-PDI与溶剂分子的溶度参数的差值(∆δ)近似说明溶剂SAs与EP-PDI分子间的相互作用程度。当∆δ的数值越小时,我们认为两种分子之间具有越强的相互作用83。根据与EP-PDI分子间作用程度的不同,我们可以将所选择的溶剂SAs划分为两类,如表4所示:第一类溶剂与 EP-PDI具有相对较弱的相互作用(即对EP-PDI溶解性较差),如DIO和ODT;第二类溶剂与EP-PDI分子间有较强相互作用(对EP-PDI的溶解度可以达到 50 mg·mL-1以上),如 CN、BT和BF。

表4 五种不同添加剂的沸点(Tb)和相应添加剂与EP-PDI的溶度参数84Table 4 Boiling points (Tb) of five different SAs and the difference in solubility parameter between SAs and EP-PDI material84.
4.1 增加低相容性共混体系共混相含量
对共混程度较低的 PTB7/EP-PDI体系,EP-PDI分子倾向于形成微米级尺寸的聚集体,进而产生大尺寸的相分离形貌。我们采用TEM照片表征薄膜的形貌,如图8所示。显然,PTB7/EP-PDI体系由于较差的相容性,EP-PDI相更倾向于形成微米级的聚集体,而不是以无定形态分散在PTB7相中。通过分别引入第二类添加剂CN、BT以及BF后,由于SAs促进EP-PDI分子扩散作用,抑制PE-PDI聚集,薄膜的相分离程度明显降低。而与之形成巨大反差的是加入 DIO、ODT等与EP-PDI相互作用弱的第一类SAs,由于其不能促进EP-PDI扩散,无法抑制EP-PDI的聚集,对薄膜相分离形貌的改善几乎没有任何帮助。从SAED谱图上可以看出,无SAs的初始薄膜中已经可以观察到明显的代表 EP-PDI的(004)、(211)晶面的衍射环57,同时还观察到了EP-PDI的π-π堆积的衍射环。也就是说在PTB7/EP-PDI 的初始共混薄膜中,EP-PDI受体组分已经形成了大量有序堆积结构。而引入CN等第二类SAs后,(004)、(012)晶面的衍射环趋于各向同性,同时随着SAs沸点的降低衍射强度明显减弱,但是 π-π堆叠的衍射环仍然存在84。

图8 不同含量异种溶剂添加剂作用下的PTB7/ EP-PDI(1 : 2)异质结薄膜透射电镜图和选区电子衍射谱图84Fig.8 TEM images and corresponding SAED patterns of BHJ films cast from PTB7/EP-PDI (1 : 2)blended films84.(a) without additive; (b) with 0.75% DIO; (c) with 0.75% ODT; (d) with 0.75% CN; (e) with 0.75% BT; (f) with 0.4% BF.
为了研究薄膜中无定型共混相与结晶纯相随SAs的变化,我们通过UV-vis吸收光谱以及GIXD谱图表征了薄膜的结晶性,如图9所示。CN等第二类SAs的引入,使对应EP-PDI的H-聚集的600 nm附近的吸收峰明显减弱85,86,与此同时,在400-550 nm之间代表EP-PDI单个分子光谱性质的0-0 (536 nm),0-1 (495 nm)以及 0-2 (466 nm)转变的三重峰明显增强。而这样的三重转变峰往往只出现在PDI类材料的稀溶液光谱中87。GIXD谱图同样显示出第二类 SAs对衍射峰的影响,对应EP-PDI的(200)、(400)晶面的衍射峰较初始薄膜明显减弱。可见第二类SAs在抑制EP-PDI聚集尺寸的同时,可以有效促进 EP-PDI无定型形态的产生,从而增加薄膜中给体/受体共混相的比例。而从薄膜吸收光谱以及GIXD谱图来看,DIO等第一类SAs的引入则几乎没有带来显著的变化,这一现象与之前薄膜形态的变化规律是一致的。通过选择第二类SAs,有效抑制EP-PDI聚集尺寸的同时增强了薄膜中共混相比例,有助于共混相与结晶纯相之间能级梯度的形成,从而促进电荷转移态的产生与分离过程。如表 5所示,薄膜性质得到优化后,器件效率由0.02%提高到1.65%84。
4.2 降低高相容性共混体系共混相含量
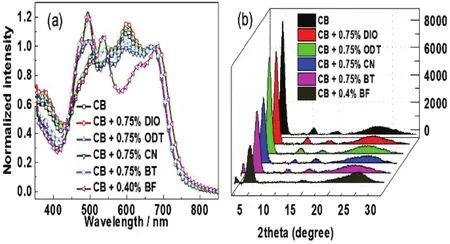
图9 不同添加剂下的PTB7/EP-PDI吸收光谱图(a)和掠入射X射线衍射谱图(b) 84Fig.9 (a) Absorption spectra and (b) GIXD patterns of the PTB7/EP-PDI blended films with different SAs84.
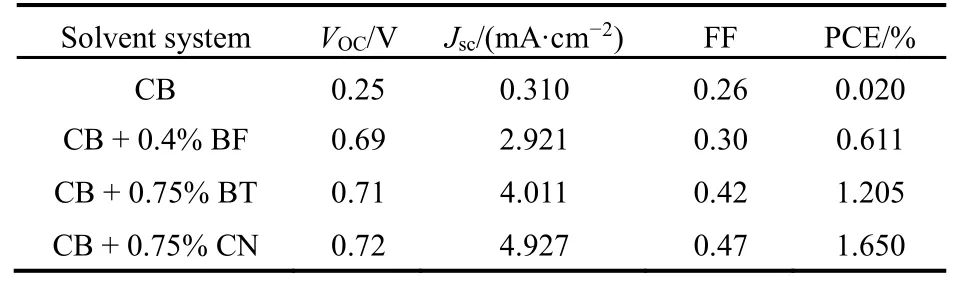
表5 不同溶剂添加剂加工的PTB7/EP-PDI (1 : 1)太阳能电池光伏特性Table 5 Photovoltaic properties of PTB7/EP-PDI (1 : 1)solar cells processed with different SAs84.
对给体/受体共混程度较高的 p- DTS(FBTTh2)2/EP-PDI体系,通过选择与EP-PDI分子具有较弱相互作用的 SAs可以有效增强EP-PDI分子之间的相互作用,构筑更多的给体/受体结晶纯相。另外,对于EP-PDI分子具有较强相互作用的 SAs,通过延长分子间作用时间,同样可以增加薄膜中给体/受体纯相比例。利用TEM表征了 p-DTS(FBTTh2)2/EP-PDI体系在不同 SAs条件下的薄膜形貌,如图10所示。与PTB7/EP-PDI体系相比,该体系给体/受体材料之间显然具有更好的相容性。共混薄膜表现为一个平整的连续薄膜,薄膜粗糙度在1 nm左右。与此同时,在SAED谱图上只能观察到较为弥散的代表 π-π堆叠的衍射环(可能来自给体或者受体),如图10a所示。当引入DIO等第一类SAs时,我们发现共混薄膜产生了明显的相分离形貌,SAED谱图上产生了EP-PDI的(004)和(211)晶面的衍射环,同时代表ππ堆积的衍射环强度也变得异常锐利,如图10b,c所示。当引入少量的CN等第二类SAs时,共混薄膜形貌与初始薄膜相近,同时薄膜变得更加平整,粗糙度进一步降低(达到 1 nm 以下),SAED上衍射环基本消失,如图10(e-g)所示。由此可见,第二类溶剂 SAs的引入不利于 p-DTS(FBTTh2)2/ EP-PDI体系相分离形貌的优化,相反会进一步增强给体与受体材料的相容性。较为异常的现象是,当CN含量进一步增大时(由0.75%提高到1.0%),共混薄膜的相分离形貌会进一步增大,同时SAED谱图上表现出与第一类SAs类似的结晶性增强的现象。

图10 不同含量异种溶剂添加剂作用下的p-DTS(FBTTh2)2/EP-PDI (1 : 1)异质结薄膜透射电镜图和选区电子衍射谱图84Fig.10 TEM images and corresponding SAED patterns of BHJ films cast from p-DTS(FBTTh2)2/EP-PDI (1 : 1)blended films 84.(a) without additive; (b) with 0.40% DIO; (c) with 0.75% ODT; (d) with 1.0% CN; (e) with 0.75% CN; (f) with 1.0% BT; (g) with 1.0% BF.
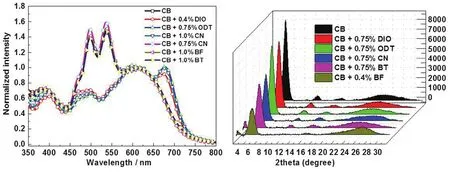
图11 不同添加剂下的p-DTS(FBTTh2)2/EP-PDI吸收光谱图(a)和掠入射X射线衍射谱图(b) 84Fig.11 (a) Absorption spectra and (b) GIXD patterns of the p-DTS(FBTTh2)2/EP-PDI blended films with different SAs 84.
为了说明 SAs对p-DTS(FBTTh2)2/EP-PDI体系共混程度以及结晶性的影响,我们对不同 SAs条件下的共混薄膜同样进行了 UV-Vis吸收光谱与GIXD谱图的表征,如图11所示。在初始薄膜的吸收光谱中,我们发现不仅EP-PDI的三重转变峰(即0-0,0-1以及0-2转变)非常明显,而且代表 p-DTS(FBTTh2)2的吸收峰只有一个(只有 610 nm附近的宽峰,而并没有观察到678 nm附近的肩峰),也就是说无论给体还是受体在该共混体系中更多地以无定形状态存在88。在GIXD谱图中,我们只观察到相对较弱的代表 EP-PDI的(200)晶面(2θ = 5.23°)的衍射峰以及 p-DTS(FBTTh2)2对应已基侧链的堆叠间距(2θ=6.18°)即(011)晶面的衍射峰。而当引入DIO等第一类SAs或者较多含量的第二类添加剂CN(1.0%)时,吸收光谱上EP-PDI的三重转变峰明显变弱,同时接近680 nm附近产生新的吸收肩峰。在 GIXD谱图上,产生了三个新的强烈的衍射峰(2θ = 4.05°,7.95°和 11.91°),分别对应p-DTS(FBTTh2)2分子中段的烷基链的堆叠间距即(001)、(002)和(003)晶面。由此可见,引入SAs后给体与受体的结晶性都得到了较为显著的提高。而当我们选择少量的第二类 SAs时,GIXD谱图上的各个衍射峰都骤然降低,尤其是BT和BF作SAs时,在GIXD谱图上几乎看不到明显的衍射峰,也就是说第二类SAs的引入不利于两组分结晶纯相的形成。对共混程度较高的p-DTS(FBTTh2)2/EP-PDI体系,由于缺少必要的固体有序结构,激子分离后形成的载流子难以有效传输到相应电极。通过选择第一类 SAs,提高给体/受体结晶纯相的比例,有效改善了电子与空穴的传输,提高了JSC,最终使器件效率由0.18%提高到2.82%,如表6所示。
本部分工作中,我们以 PTB7/EP-PDI与p-DTS(FBTTh2)2/EP-PDI为例研究了非富勒烯共混体系中共混相、给体结晶纯相与受体结晶纯相三相组成的重要作用,并利用选择性SAs手段实现了活性层三相模型的定向优化。对于共混程度较差的 PTB7/EP-PDI体系,引入与 EP-PDI分子作用力较强的溶剂SAs可有效抑制EP-PDI聚集尺寸,增强无定形共混相的比例。对于共混程度较高的 p-DTS(FBTTh2)2/EP-PDI体系,引入与EP-PDI分子作用力较弱的溶剂 SAs可促进EP-PDI结晶,从而形成更多结晶纯相。

表6 不同溶剂添加剂加工的F-DTS/EP-PDI (1 : 2)太阳能电池光伏特性Table 6 Photovoltaic properties of F-DTS/EP-PDI (1 : 2)solar cells processed with different SAs84.
5 结论与展望
非富勒烯太阳能电池PCE的突飞猛进,源于器件结构设计、材料合成及形貌优化。本文从凝聚态结构调控角度,着重分析了基于 PDI类非富勒烯受体材料共混体系相分离结构、相区尺寸及共混相含量调控。指出,给受体比例及分子扩散能力是决定相分离结构的主要因素,通过调节比例及热退火温度获得纳米级互穿网络结构是制备高性能器件的前提。在此基础上,通过增加溶剂-溶质分子间相互作用/引入给受体间电荷转移相互作用实现了低相容性/高相容性共混体系相区尺寸的调控。最后,通过调节溶剂与PDI分子间δ,实现了活性层中共混相含量的调节,指出对于低相容性共混体系应当选择与PDI分子δ差异小的溶剂,而对于高相容性共混体系则应当选择与 PDI分子δ差异大的溶剂。
然而,非富勒烯共混体系相分离结构复杂,尤其是以分子级别混合的共混相对器件光物理过程的影响尚不清晰。因此,在相分离结构、相区尺寸及共混相含量优化的基础上,寻求热力学及动力学手段进一步调节共混相中给体分子与受体分子的含量,建立相关含量与能量损失、CT态分离效率及载流子传输过程中双分子复合间关系,深入理解器件工作过程中的光物理过程,是进一步优化器件性能的突破点!另外,富勒烯分子可近似看作球形,为各向同性分子;而非富勒烯分子为各向异性分子,其取向方式分为三种,包含edge-on、flat-on及face-on取向。大量研究表明,当界面处给体分子与受体分子取向一致时,给受体分子四极矩诱导产生的电势方向一致,可最大程度上促进CT态分离;反之,当界面处给受体分子取向不一致时,则不利于CT态分离89,90。对于非富勒烯共混体系而言,给受体分子介电常数均较小,界面处形成的CT态易转变为三线态或复合至基态。侯剑辉课题组91,92研究表明通过增加分子单元结构的共轭面积可以促进界面处给受体分子采取相同取向,而这种分子取向结构十分有助于提升CT态分离效率,进一步证实了界面处分子取向研究的重要意义。因此,如何通过分子设计或凝聚态结构调控手段实现界面处分子取向一致,进一步提高CT态分离效率,也是非富勒烯体系值得进一步研究的突破点。
(1) He, Z. C.; Zhong, C. M.; Su, S. J.; Xu, M.; Wu, H. B.; Cao, Y. Nat.Photon. 2012, 6, 591. doi: 10.1038/Nphoton.2012.190
(2) You, J. B.; Dou, L. T.; Yoshimura, K.; Kato, T.; Ohya, K.; Moriarty, T.; Emery, K.; Chen, C. C.; Gao, J.; Li, G.; Yang, Y. Nat. Commun.2013, 4, 1446. doi: 10.1038/ncomms2411
(3) Li, G. F.; Zuo, Y.; Chen, L.; Zhang, J. D.; Yan, D. H.; Qin, D. S. Acta Polym. Sin. 2013, 13, 183. [李贵芳, 左阳, 陈磊, 张吉东, 闫东航, 秦大山. 高分子学报, 2013, 13, 183.] doi: 10.3724/SP.J.1105.2013.12190
(4) Feng, Y.; Su, Z. X.; Sun, L.; liu, S. G.; Diao, K. S.; Zhang, W. M. Acta Polym. Sin. 2014, 1613. [冯宇, 苏智兴, 孙丽, 刘绍刚, 刁开盛, 张卫民. 高分子学报, 2014, 1613.] doi: 10.11777/j.issn1000-3304.2014.14110 (5) Li, Y. F. Acta Phys. -Chim. Sin. 2017, 33, 447. [李永舫. 物理化学学报, 2017, 33, 447.] doi: 10.3866/PKU.WHXB201702132 (6) Luo, H. W.; Liu, Z. T. Chin.Chem.Lett. 2016, 27, 1283. doi: 10.1016/j.cclet.2016.07.003
(7) Shen, X. X.; Han, G. C.; Yi, Y. P. Chin.Chem.Lett. 2016, 27, 1453. doi: 10.1016/j.cclet.2016.05.030
(8) Qin, Y. P.; Chen, Y.; Cui, Y.; Zhang, S. Q.; Yao, H. F.; Huang, J.; Li, W .N.; Zheng, Z.; Hou, J. H. Adv. Mater. 2017, 29, 1606340 doi: 10.1002/adma.201606340
(9) Zhao, W. C.; Li, S. S.; Zhang, S. Q.; Liu, X. Y.; Hou, J. H. Adv.Mater. 2017, 29, 1604359. doi: 10.1002/adma.201604059
(10) Kim, Y.; Cook, S.; Tuladhar, S. M.; Choulis, S. A.; Nelson, J.; Durrant, J. R.; Bradley, D. D. C.; Giles, M.; Mcculloch, I.; Ha, C. S.; Ree, M. Nature Mater. 2006, 5, 197. doi: 10.1038/nmat1574
(11) Bijleveld, J. C.; Zoombelt, A. P.; Mathijssen, S. G. J.; Wienk, M. M.; Turbiez, M.; de Leeuw, D. M.; Janssen, R. A. J. J. Am. Chem. Soc.2009, 131, 16616. doi: 10.1021/ja907506r
(12) Reshma, L.; Santhakumar, K. Org. Electron. 2017, 47, 35. doi: 10.1016/j.orgel.2017.05.002
(13) Bi, P. Q.; Zheng, F.; Yang, X. Y.; Niu, M. S.; Feng, L.; Qin, W.; Hao, X. T. J. Mater. Chem. A 2017, 5, 12120. doi: 10.1039/c7ta01557g
(14) Chen, S. S.; Yao, H. T.; Li, Z. K.; Awartani, O. M.; Liu, Y. H.; Wang, Z.; Yang, G. F.; Zhang, J. Q.; Ade, H.; Yan, H. Adv. Energy Mater. 2017, 7, 1602304. doi: 10.1002/aenm.201602304
(15) Baran, D.; Ashraf, R. S.; Hanifi, D. A.; Abdelsamie, M.; Gasparini, N.; Rohr, J. A.; Holliday, S.; Wadsworth, A.; Lockett, S.; Neophytou, M.; Emmott, C. J.; Nelson, J.; Brabec, C. J.; Amassian, A.; Salleo, A.; Kirchartz, T.; Durrant, J. R.; McCulloch, I. Nat.Mater. 2017, 16, 363. doi: 10.1038/nmat4797
(16) Zhao, W. C.; Li, S. S.; Yao, H. F.; Zhang, S. Q.; Zhang, Y.; Yang, B.; Hou, J. H. J. Am. Chem. Soc. 2017, 139, 7148. doi: 10.1021/jacs.7b02677
(17) Tang, C. W. Appl. Phys. Lett. 1986, 48, 183. doi: 10.1063/1.96937
(18) Wei, Z. X. Acta Phys. -Chim. Sin. 2017, 33, 2119. [魏志祥. 物理化学学报, 2017, 33, 2119.] doi: 10.3866/PKU.WHXB201706141
(19) Mazzio, K. A.; Luscombe, C. K. Chem. Soc. Rev. 2015, 44, 78. doi: 10.1039/c4cs00227j
(20) Nielsen, C. B.; Holliday, S.; Chen, H. Y.; Cryer, S. J.; McCulloch, I. Acc. Chem. Res. 2015, 48, 2803. doi: 10.1021/acs.accounts.5b00199
(21) Li, Y. F. Acta Phys. -Chim. Sin. 2017, 33, 268. [李永舫. 物理化学学报, 2017, 33, 268.] doi: 10.3866/PKU.WHXB201701031
(22) Min, J.; Kwon, O. K.; Cui, C. H.; Park, J. -H.; Wu, Y.; Park, S. Y.; Li, Y. F.; Brabec, C. J. J. Mater. Chem. A 2016, 4, 14234. doi: 10.1039/c6ta05303c
(23) Fu, Y. Y.; Wang, B.; Qu, J. F.; Wu, Y.; Ma, W.; Geng, Y. H.; Han, Y. C.; Xie, Z. Y. Adv. Funct. Mater. 2016, 26, 5922. doi: 10.1002/adfm.201601880
(24) Bloking, J. T.; Han, X.; Higgs, A. T.; Kastrop, J. P.; Pandey, L.; Norton, J. E.; Risko, C.; Chen, C. E.; Brédas, J. -L.; McGehee, M. D.; Sellinger, A. Chem. Mater. 2011, 23, 5484. doi: 10.1021/cm203111k
(25) Zhou, Y.; Ding, L.; Shi, K.; Dai, Y. Z.; Ai, N.; Wang, J.; Pei, J. Adv.Mater. 2012, 24, 957. doi: 10.1002/adma.201103927
(26) Zhou, Y.; Dai, Y. Z.; Zheng, Y. Q.; Wang, X. Y.; Wang, J. Y.; Pei, J. Chem. Commun. 2013, 49, 5802. doi: 10.1039/c3cc41803k
(27) Winzenberg, K. N.; Kemppinen, P.; Scholes, F. H.; Collis, G. E.; Shu, Y.; Singh, T. B.; Bilic, A.; Forsyth, C. M.; Watkins, S. E. Chem. Commun. 2013, 49, 6307. doi: 10.1039/c3cc42293c
(28) Lin, Y. Z.; Li, Y. F.; Zhan, X. W. Adv. Energy Mater. 2013, 3, 724. doi: 10.1002/aenm.201200911
(29) Lin, Y. Z.; Zhang, Z. G.; Bai, H. T.; Wang, J. Y.; Yao, Y. H.; Li, Y. F.; Zhu, D. B.; Zhan, X. W. Energy Environ. Sci. 2015, 8, 610. doi: 10.1039/c4ee03424d
(30) Lin, Y. Z.; Wang, J. Y.; Zhang, Z. G.; Bai, H. T.; Li, Y. F.; Zhu, D. B.; Zhan, X. W. Adv. Mater. 2015, 27, 1170. doi: 10.1002/adma.201404317
(31) Zhao, W. C.; Qian, D. P.; Zhang, S. Q.; Li, S. S.; Inganas, O.; Gao, F.; Hou, J. H. Adv. Mater. 2016, 28, 4734. doi: 10.1002/adma.201600281
(32) Rajaram, S.; Shivanna, R.; Kandappa, S. K.; Narayan, K. S. J. Phys.Chem. Lett. 2012, 3, 2405. doi: 10.1021/jz301047d
(33) Zhang, X.; Zhan, C. L.; Yao, J. N. Chem. Mater. 2015, 27, 166. doi: 10.1021/cm504140c
(34) Zang, Y.; Li, C. Z.; Chueh, C. C.; Williams, S. T.; Jiang, W.; Wang, Z. H.; Yu, J. S.; Jen, A. K. Adv. Mater. 2014, 26, 5708. doi: 10.1002/adma.201401992
(35) Zhong, Y.; Trinh, M. T.; Chen, R.; Wang, W.; Khlyabich, P. P.; Kumar, B.; Xu, Q.; Nam, C. Y.; Sfeir, M. Y.; Black, C.; Steigerwald, M. L.; Loo, Y. L.; Xiao, S.; Ng, F.; Zhu, X. Y.; Nuckolls, C. J. Am.Chem. Soc. 2014, 136, 15215. doi: 10.1021/ja5092613
(36) Chiu, M. Y.; Jeng, U. S.; Su, M. S.; Wei, K. H. Macromolecules 2010, 43, 428. doi: 10.1021/ma901895d
(37) Alam, M. M.; Tonzola, C. J.; Jenekhe, S. A. Macromolecules 2003,36, 6577. doi: 10.1021/ma0346299
(38) Sepe, A.; Rong, Z. X.; Sommer, M.; Vaynzof, Y.; Sheng, X. Y.; Muller-Buschbaum, P.; Smilgies, D. M.; Tan, Z. K.; Yang, L. F., R. H.; Steiner, U.; Huttner, S. ENERG ENVIRON SCI 2014, 7, 1725. doi: 10.1039/c3ee44125c
(39) Wolfer, P.; Schwenn, P. E.; Pandey, A. K.; Fang, Y.; Stingelin, N.; Burn, P. L.; Meredith, P. J. Mater. Chem. A 2013, 1, 5989. doi: 10.1039/c3ta10554g
(40) Ren, G. Q.; Ahmed, E.; Jenekhe, S. A. Adv. Energy Mater. 2011, 1, 946. doi: 10.1002/aenm.201100285
(41) Ye, L.; Jiang, W.; Zhao, W. C.; Zhang, S. Q.; Qian, D. P.; Wang, Z. H.; Hou, J. H. Small 2014, 10, 4658. doi: 10.1002/smll.201401082
(42) Sharenko, A.; Proctor, C. M.; van der Poll, T. S.; Henson, Z. B.; Nguyen, T. Q.; Bazan, G. C. Adv. Mater. 2013, 25, 4403. doi: 10.1002/adma.201301167
(43) Ye, T.; Singh, R.; Butt, H. J.; Floudas, G.; Keivanidis, P. E. ACS Appl. Mat. Interfaces 2013, 5, 11844. doi: 10.1021/am4035416
(44) Singh, R.; Giussani, E.; Mróz, M. M.; Di Fonzo, F.; Fazzi, D.; Cabanillas-González, J.; Oldridge, L.; Vaenas, N.; Kontos, A. G.; Falaras, P.; Grimsdale, A. C.; Jacob, J.; Müllen, K.; Keivanidis, P. E. Org. Electron. 2014, 15, 1347. doi: 10.1016/j.orgel.2014.03.044
(45) Fu, Y. Y.; Yang, Q. Q.; Deng, Y. F.; Jiang, W.; Wang, Z. H.; Geng, Y. H.; Xie, Z. Y. Org. Electron. 2015, 18, 24. doi: 10.1016/j.orgel.2015.01.008
(46) Liang, Q. J.; Han, J.; Song, C. P.; Wang, Z. Y.; Xin, J. M.; Yu, X. H.; Xie, Z. Y.; Ma, W.; Liu, J. G.; Han, Y. C. J. Mater. Chem. C 2017, 5, 6842. doi: 10.1039/c7tc01763d
(47) Kamm, V.; Battagliarin, G.; Howard, I. A.; Pisula, W.; Mavrinskiy, A.; Li, C.; Müllen, K.; Laquai, F. Adv. Energy Mater. 2011, 1, 297. doi: 10.1002/aenm.201000006
(48) Zhao, J. B.; Li, Y. K.; Lin, H. R.; Liu, Y. H.; Jiang, K.; Mu, C.; Ma, T. X.; Lin Lai, J. Y.; Hu, H. W.; Yu, D. M.; Yan, H. Energy Environ. Sci. 2015, 8, 520. doi: 10.1039/c4ee02990a
(49) Zhang, X.; Lu, Z. H.; Ye, L.; Zhan, C. L.; Hou, J. H.; Zhang, S. Q.; Jiang, B.; Zhao, Y.; Huang, J. H.; Zhang, S. L.; Liu, Y.; Shi, Q.; Liu, Y. Q.; Yao, J. N. Adv. Mater. 2013, 25, 5791. doi: 10.1002/adma.201300897
(50) Lu, Z. H.; Jiang, B.; Zhang, X.; Tang, A. L.; Chen, L. L.; Zhan, C. L.; Yao, J. N. Chem. Mater. 2014, 26, 2907. doi: 10.1021/cm5006339
(51) Lin, Y. Z.; Wang, Y. F.; Wang, J. Y.; Hou, J. H.; Li, Y. F.; Zhu, D. B.; Zhan, X. W. Adv. Mater. 2014, 26, 5137. doi: 10.1002/adma.201400525
(52) Liu, Y. H.; Mu, C.; Jiang, K.; Zhao, J. B.; Li, Y. K.; Zhang, L.; Li, Z. K.; Lai, J. Y. L.; Hu, H. W.; Ma, T. X.; Hu, R. R.; Yu, D. M.; Huang, X. H.; Tang, B. Z.; Yan, H. Adv. Mater.2015, 27, 1015. doi: 10.1002/adma.201404152
(53) Chen, W. Q.; Yang, X.; Long, G. K.; Wan, X. J.; Chen, Y. S.; Zhang, Q. C. J. Mater. Chem. C 2015, 3, 4698. doi: 10.1039/c5tc00865d
(54) Sun, D.; Meng, D.; Cai, Y. H.; Fan, B. B.; Li, Y.; Jiang, W.; Huo, L. J.; Sun, Y. M.; Wang, Z. H. J. Am. Chem. Soc. 2015,137, 11156. doi: 10.1021/jacs.5b06414
(55) Rajaram, S.; Armstrong, P. B.; Kim, B. J.; ´chet, J. M. J. F.Chem. Mater. 2009, 21, 1775. doi: 10.1021/cm900911x
(56) Li, M. G.; Wang, L.; Liu, J. G.; Zhou, K.; Yu, X. H.; Xing, R. B.; Geng, Y. H.; Han, Y. C. Phys. Chem. Chem. Phys. 2014,16, 4528. doi: 10.1039/c3cp55075c
(57) Wiatrowski, M.; Dobruchowska, E.; Maniukiewicz, W.; Pietsch, U.; Kowalski, J.; Szamel, Z.; Ulanski, J. Thin Solid Films 2010, 518, 2266. doi: 10.1016/j.tsf.2009.08.037
(58) Yang, C.; Orfino, F. P.; Holdcroft, S. Macromolecules 1996,29, 6510. doi: 10.1021/ma9604799
(59) Wu, F. C.; Hsu, S. W.; Cheng, H. L.; Chou, W. Y.; Tang, F. C. J. Phys. Chem. C 2013, 117, 8691. doi: 10.1021/jp400849x
(60) Liu, J. G.; Chen, L.; Gao, B. R.; Cao, X. X.; Han, Y. C.; Xie, Z. Y.; Wang, L. X. J. Mater. Chem. A 2013, 1, 6216. doi: 10.1039/c3ta10629b
(61) Liang, Y. Y.; Xu, Z.; Xia, J. B.; Tsai, S. -T.; Wu, Y.; Li, G.; Ray, C.; Yu, L. P. Adv. Mater. 2010, 22, E135. doi: 10.1002/adma.200903528
(62) Chu, T. Y.; Lu, J.; Beaupre, S.; Zhang, Y.; Pouliot, J. R.; Wakim, S.; Zhou, J.; Leclerc, M.; Li, Z.; Ding, J.; Tao, Y. J. Am. Chem. Soc. 2011, 133, 4250. doi: 10.1021/ja200314m
(63) Guo, X.; Cui, C. H.; Zhang, M. J.; Huo, L. J.; Huang, Y.; Hou, J. H.; Li, Y. F. Energy Environ. Sci. 2012, 5, 7943. doi: 10.1039/c2ee21481d
(64) Howard, I. A.; Laquai, F. d. r.; Keivanidis, P. E.; Friend, R. H.; Greenham, N. C. J. Phys. Chem. C 2009, 113, 21225. doi: 10.1021/jp907633g
(65) Yang, B.; Zhang, S. Q.; Chen, Y.; Cui, Y.; Liu, D. L.; Yao, H. F.; Zhang, J. Q.; Wei, Z. X.; Hou, J. H. Macromolecules 2017, 50, 1453. doi: 10.1021/acs.macromol.6b02733
(66) Zhang, S. Q.; Hou, J. H. Acta Phys. -Chim. Sin. 2017, 33, 0001. [张少青, 侯剑辉. 物理化学学报, 2017, 33, 0001.] doi: 10.3866/PKU.WHXB201706161
(67) Li, M. G.; Liang, Q. J.; Zhao, Q. Q.; Zhou, K.; Yu, X. H.; Xie, Z. Y.; Liu, J. G.; Han, Y. C. J. Mater. Chem. C 2016, 4, 10095. doi: 10.1039/c6tc03061k
(68) Burke, T. M.; McGehee, M. D. Adv. Mater. 2014, 26, 1923. doi: 10.1002/adma.201304241
(69) Westacott, P.; Tumbleston, J. R.; Shoaee, S.; Fearn, S.; Bannock, J. H.; Gilchrist, J. B.; Heutz, S.; deMello, J.; Heeney, M.; Ade, H.; Durrant, J.; McPhail, D. S.; Stingelin, N. Energy Environ. Sci. 2013, 6, 2756. doi: 10.1039/c3ee41821a
(70) Collins, B. A.; Tumbleston, J. R.; Ade, H. J. Phys. Chem.Lett.2011, 2, 3135. doi: 10.1021/jz2014902
(71) Groves, C. Energy Environ. Sci. 2013, 6, 1546. doi: 10.1039/c3ee24455e
(72) Sweetnam, S.; Graham, K. R.; Ngongang Ndjawa, G. O.; Heumuller, T.; Bartelt, J. A.; Burke, T. M.; Li, W.; You, W.; Amassian, A.; McGehee, M. D. J. Am. Chem. Soc. 2014, 136, 14078. doi: 10.1021/ja505463r
(73) Bloking, J. T.; Giovenzana, T.; Higgs, A. T.; Ponec, A. J.; Hoke, E. T.; Vandewal, K.; Ko, S.; Bao, Z.; Sellinger, A.; McGehee, M. D. Adv. Energy Mater. 2014, 4, 1301426. doi: 10.1002/aenm.201301426
(74) Jamieson, F. C.; Domingo, E. B.; McCarthy-Ward, T.; Heeney, M.; Stingelin, N.; Durrant, J. R. Chem. Sci. 2012, 3, 485. doi: 10.1039/c1sc00674f
(75) Shaw, P. E.; Wolfer, P.; Langley, B.; Burn, P. L.; Meredith, P. J. Phys. Chem. C 2014, 118, 13460. doi: 10.1021/jp503150u
(76) Zhou, E.; Cong, J.; Hashimoto, K.; Tajima, K. Adv. Mater.2013, 25, 6991. doi: 10.1002/adma.201303170
(77) Vongsaysy, U.; Pavageau, B.; Wantz, G.; Bassani, D. M.; Servant, L.; Aziz, H. Adv. Energy Mater. 2014, 4, 1300752. doi: 10.1002/aenm.201300752
(78) Liu, J. G. ;Shao, S. Y.; Wang, H. F.; Zhao, K.; Xue, L .J.; Gao, X.; Xie, Z. Y.; Han, Y. C. Org. Electron. 2010, 11, 775. doi: 10.1016/j.orgel.2010.01.017
(79) Dang, M. T.; Wuest, J. D. Chem. Soc. Rev. 2013, 42, 9105. doi: 10.1039/c3cs35447d
(80) Lee, J. K.; Ma, W. L.; Brabec, C. J.; Yuen, J.; Moon, J. S.; Kim, J. Y.; Lee, K.; Bazan, G. C.; Heeger, A. J. J. Am. Chem.Soc. 2008, 130, 3619. doi: 10.1021/ja710079w
(81) Graham, K. R.; Wieruszewski, P. M.; Stalder, R.; Hartel, M. J.; Mei, J.; So, F.; Reynolds, J. R. Adv. Funct. Mater. 2012,22, 4801. doi: 10.1002/adfm.201102456
(82) Su, M. S.; Kuo, C. Y.; Yuan, M. C.; Jeng, U. S.; Su, C. J.; Wei, K. H. Adv. Mater. 2011, 23, 3315. doi: 10.1002/adma.201101274
(83) Salim, T.; Wong, L. H.; Bräuer, B.; Kukreja, R.; Foo, Y. L.; Bao, Z.; Lam, Y. M. J. Mater. Chem. 2011, 21, 242. doi: 10.1039/c0jm01976c
(84) Li, M. G.; Liu, J. G.; Cao, X. X.; Zhou, K.; Zhao, Q. Q.; Yu, X. H.; Xing, R. B.; Han, Y. C. Phys. Chem. Chem. Phys.2014, 16, 26917. doi: 10.1039/c4cp04161e
(85) Wurthner, F. Chem. Commun. 2004, 1564. doi: 10.1039/b401630k
(86) Chen, Z.; Stepanenko, V.; Dehm, V.; Prins, P.; Siebbeles, L. D.; Seibt, J.; Marquetand, P.; Engel, V.; Wurthner, F. Chem.Eur. J. 2007, 13, 436. doi: 10.1002/chem.200600889
(87) Balakrishnan, K.; Datar, A.; Oitker, R.; Chen, H.; Zuo, J.; Zang, L. J. Am. Chem. Soc. 2005, 127, 10496. doi: 10.1021/ja052940v
(88) Love, J. A.; Proctor, C. M.; Liu, J.; Takacs, C. J.; Sharenko, A.; van der Poll, T. S.; Heeger, A. J.; Bazan, G. C.; Nguyen, T. -Q. Adv. Funct. Mater. 2013, 23, 5019. doi: 10.1002/adfm.201300099
(89) Verlaak, S.; Beljonne, D.; Cheyns, D.; Rolin, C.; Linares, M.; Castet, F.; Cornil, J.; Heremans, P. Adv. Funct. Mater. 2009,19, 3809. doi: 10.1002/adfm.200901233
(90) Ye, L.; Jiao, X. C.; Zhou, M.; Zhang, S. Q.; Yao, H. F.; Zhao, W. C.; Xia, A. D.; Ade, H.; Hou, J. H. Adv. Mater. 2015, 27, 6046. doi: 10.1002/adma.201503218
(91) Zhang, H.; Ye, L.; Hou, J. H. Polym. Int. 2015, 64, 957. doi: 10.1002/pi.4895
(92) Ye, L.; Jiao, X. C.; Zhang, H.; Li, S. S.; Yao, H. F.; Ade, H.; Hou, J. H. Macromolecules 2015, 48, 7156. doi: 10.1021/acs.macromol.5b01537
猜你喜欢
学与玩(2022年12期)2023-01-11 06:39:22
中国化妆品(2019年4期)2019-11-20 01:47:53
同煤科技(2019年1期)2019-05-16 01:46:22
橡塑技术与装备(2018年10期)2018-05-18 18:16:16
化工管理(2017年12期)2017-05-12 08:38:28
厦门大学学报(自然科学版)(2016年4期)2016-08-04 08:18:49
中国塑料(2016年9期)2016-06-13 03:18:50
中国塑料(2015年7期)2015-10-14 01:02:40
物理化学学报(2015年5期)2015-02-28 17:34:55
火炸药学报(2014年1期)2014-03-20 13:17:24
