
3-季碳氧化吲哚合成及抗肿瘤活性研究进展
2018-05-03 06:58李楠柯梁光平丁丽娜陈东林聂昌平刘雄利
铜仁学院学报 2018年3期
杨 俊,李楠柯,梁光平,丁丽娜,陈东林,聂昌平*,刘雄利,周 英
( 1.遵义医药高等专科学校, 贵州 遵义 563006;2.贵州大学 贵州省中药民族药创制工程中心,贵州 贵阳 550025)
0.引言
先导化合物的主动筛选是对天然化合物及人工合成化合物进行生物活性的筛选,对活性较好的物质结构进行改造以提高活性及药代动力学参数等。先导化合物通常具有较新颖的化学结构及良好的理化性质。药物开发过程中,先导化合物的确定直接影响整个研究开发过程是否会成功。3-季碳氧化吲哚骨架广泛地存在于活性化合物中,如trychnofoline[1]、MI-77301[2]、NITD 609[3]、SpirotryprostatinA 及Spirotryprostatin B[4]、Gelsemine[5]、(+)-elacomine[6],结构如图1所示。
3-季碳氧化吲哚衍生物具有抗肿瘤、抗疟疾、抗抑郁、抗菌消炎等广泛的生物活性,其中抗肿瘤活性尤为突出。因为3-季碳氧化吲哚以其特有的生物活性和空间结构的特殊性,使其具有成为药物的潜力,引起了广泛的关注。如何简单有效地合成这一类分子的方法具有重要的意义,文中主要综述了合成3-季碳氧化吲哚的方法及其衍生物的抗肿瘤活性。
1.3-季碳氧化吲哚的主要合成方法
3-季碳氧化吲哚衍生物由于其特殊的生物活性,一直是有机化学家合成的热点,合成3-季碳氧化吲哚的方法主要有以下几种:C-H活化反应、Morita-Baylis-Hillman反应、Micheal加成反应、3+2环加成、aldol反应、mannich反应、串联反应、共轭加成等。
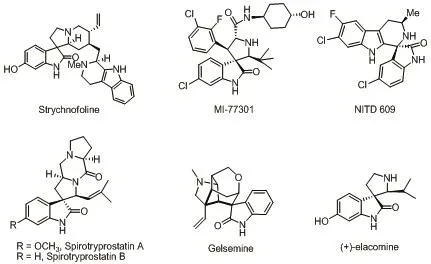
图1 具有3-季碳氧化吲哚骨架的活性化合物Fig.1 Active compounds with 3-quaternary carbon oxindole skeleton
1.1.通过C-H活化反应构建3-季碳氧化吲哚骨架
Hartwig和Buchwald等报道了通过C-H活化反应构建3-季碳氧化吲哚的诸多方法,但是这些反应都需要底物有特定的取代基,才可以发生芳基化或烷基化反应得到3-季碳氧化吲哚,不然不会发生反应。所以Yi-Xia Jia等[7]总结了这类反应的不足之处,提出了一种底物适用性较广的方法,通过Csp2-H和Csp3-H直接偶联的C-H活化历程合成3-季碳氧化吲哚的方法,经过反应条件优化得到,N,N-二甲基甲酰胺为溶剂,氧化剂为CuCl2,tBuONa作碱,温度为110℃,反应5 h,反应产率最高达97%。机理及合成路线如图2所示。
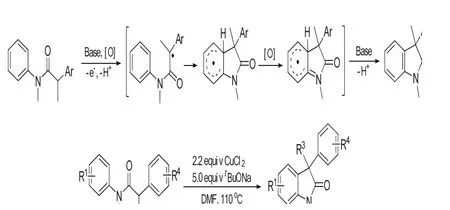
图2 通过C-H活化反应构建3-季碳氧化吲哚Fig.2 Construction of 3-quaternary carbon oxindole skeleton by C-H activations
Moody C L等[8]报道了直接用C-H活化形成3-季碳螺环氧化吲哚,用经济环保的一水合物醋酸铜(II)(10 mol%)作为催化剂,溶剂为Mesitylene,温度为170℃,最初在此反应条件下原料分解导致产率较低,后来通过泵入空气后解决这个问题。通过这个方法得到的螺环氧化吲哚,可再经过两步反应衍生得到天然化合物horsfiline,为全合成horsfiline提供了一个方法。反应如图3所示。
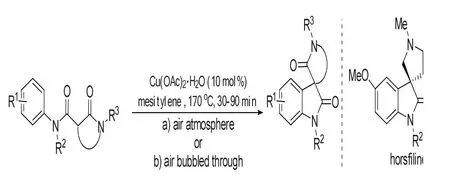
图3 通过C-H活化反应构建3-季碳氧化吲哚Fig.3 Construction of 3-quaternary carbon oxindole skeleton by C-H activations
1.2.通过Morita-Baylis-Hillman反应构建3-季碳氧化吲哚
Yun L L等[9]首次报道了靛红作为亲电试剂,亲核试剂为丙烯醛,在CH2Cl2为溶剂,β-ICD催化下,发生不对称Morita-Baylis-Hillman反应,如图4所示。该反应的底物拓展性良好,各种不同取代吲哚醌均能够与丙烯醛发生反应,收率最高达97%,ee值最高达99%,生成3-羟基季碳氧化吲哚衍生物。靛红与丙烯酸酯发生MBH生成的碳酸酯可进一步发生反应,Cui H L等[10]报道了靛红MBH碳酸酯在路易斯碱催化下,发生了不对称烯丙基烷基化反应,得到的产物可进一步发生分子内迈克尔反应生成螺环氧化吲哚。
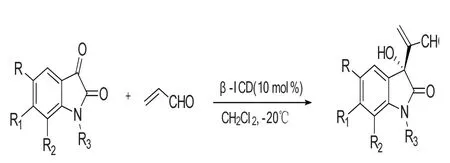
图4 通过Morita-Baylis-Hillman反应构建3-季碳氧化吲哚Fig.4 4Construction of 3-quaternary carbon oxindole skeleton by Morita-Baylis-Hillman reactions
1.3.通过Micheal加成反应构建3-季碳氧化吲哚
许多文献报道了3-取代氧化吲哚作为亲核试剂,与α,β不饱和醛酮发生Micheal加成反应来构建3-季碳氧化吲哚。Maruoka课题组[11]于2009年报道了用手性相转移催化剂膦盐3a催化3-苯基氧化吲哚与α,β不饱和醛酮发生Micheal加成反应,如图5所示。该反应具有收率高(91%~99%),对映体过量值高(90%~99%)等特点,且生成的产物可进一步衍生化,经过2~3步反应衍生为毒扁豆碱及其类似物的骨架结构。
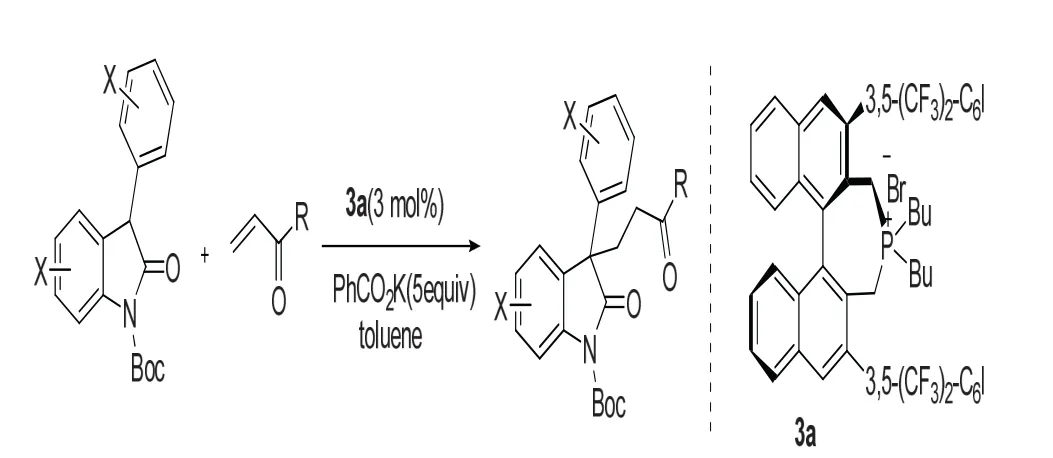
图5 通过Micheal加成反应构建3-季碳氧化吲哚Fig.5 Construction of 3-quaternary carbon ox-indole skeleton by Michael addition reactions
Miao Ding等[12]报道了奎宁丁催化3-芳基氧化吲哚与硝基烯发生Micheal加成反应,如图6所示,反应条件筛选结果为:催化剂奎宁丁为10 mol%,丙酮为溶剂,0℃下反应,该反应产率高,非对映选择性中等到好,且当使用手性奎宁丁硫脲衍生物催化时,对该反应具有一定的手性诱导作用。
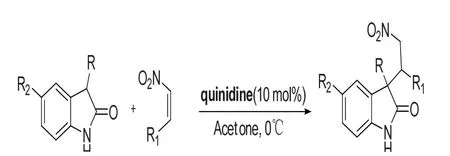
图6 通过Micheal加成反应构建3-季碳氧化吲哚Fig.6 Construction of 3-quaternary carbon oxindole skeleton by Michael addition reactions
1.4.通过3+2环加成反应构建3-季碳氧化吲哚
Liu Xiongwei等[13]报道了一种简单高效的构建3-螺环氧化吲哚的方法,他们使用吲哚醌与1-(3-甲基-4-硝基异恶唑)苯乙烯和氨基酸,在80℃下,乙腈作为溶剂,发生3+2环合得到目标化合物,如图7所示。反应首先是吲哚醌与氨基酸形成1,3偶极子然后再对1-(3-甲基-4-硝基异恶唑)苯乙烯的双键进行加成,共使用了脯氨酸,硫代脯氨酸,肌氨酸,共计得到40个3-螺环氧化吲哚,产率最高达90%,非对映选择性大于20:1。并采用MTT法对合成的这40个化合物进行体外抗肿瘤活性测试,分别对人前列腺癌细胞(PC-3)、白血病细胞(K562)、肺癌细胞(A549)进行细胞毒实验,结果表明,大多数化合物对人白血病细胞(K562)抑制作用较好,其中化合物4a(IC50=10.7 μM)活性最佳,优于阳性对照药顺铂(IC50=23.2 μM)。
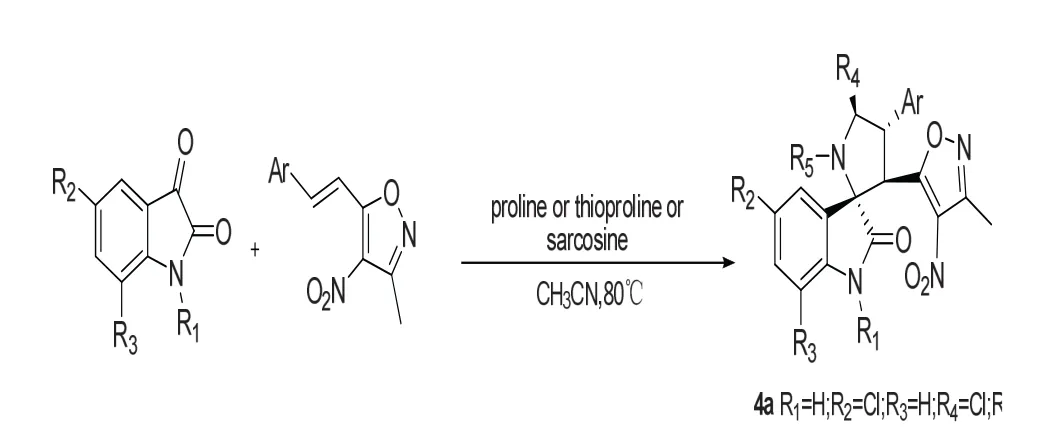
图7 通过3+2环加成反应构建3-季碳氧化吲哚Fig.7 Construction of 3-quaternary carbon oxindole skeleton by 3+2 cycloadditions
Pandi Dhanalakshmid等[14]报道了吲哚醌与4b和脯氨酸发生3+2环合反应,该反应不仅具有良好的化学选择性,还具有良好的区域和立体选择性,4b中具有两个双键,分别为双键A和双键B,即使吲哚醌和脯氨酸都是过量的,反应几乎全部发生在双键A。并提出了机理是吲哚醌首先和脯氨酸发生反应脱水,然后脱二氧化碳生成1,3偶极子,然后再和4b发生加成反应。反应如图8所示。
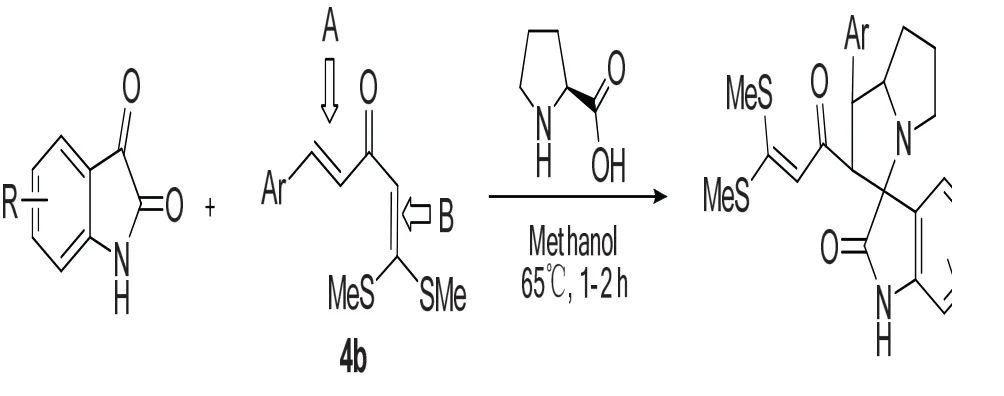
图8 通过3+2环加成反应构建3-季碳氧化吲哚Fig.8 Construction of 3-quaternary carbon oxindole skeleton by 3+2 cycloadditions
Filatov,A.S等[15]报道了吲哚醌、α-氨基酸、环丙烯在甲醇为溶剂,回流4 h后,一锅法得到3-螺环氧化吲哚,该方法可得到具有环丙烷并环结构的3-螺环氧化吲哚,产率较好,具有较好的对应选择性。反应机理:首先是吲哚醌和α-氨基酸形成亚胺后,再和取代环丙烯加成,随后进行了体外的抗肿瘤活性测定,结果表示,该类化合物具有一定的抗肿瘤活性。反应图9所示。
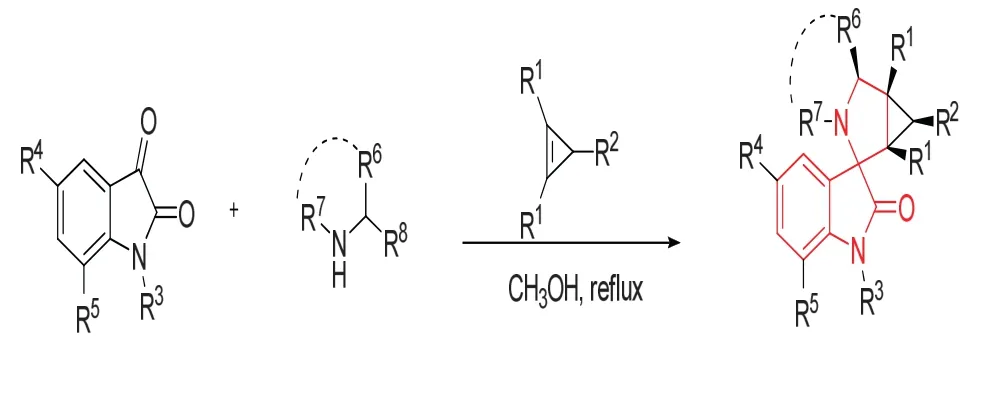
图9 通过3+2环加成反应构建3-季碳氧化吲哚Fig.9 Construction of 3-quaternary carbon oxindole skeleton by 3+2 cycloadditions
1.5.通过mannich反应构建3-季碳氧化吲哚
2008年,Xu Tian课题组[16]首次报道了3-取代的氧化吲哚的不对称mannich反应,填补了通过mannich反应构建3位具有手性碳原子氧化吲哚的空白,使用3-取代氧化吲哚和N-Boc亚胺在双功能硫脲催化剂6a的催化下,通过不对称mannich反应构建3-季碳氧化吲哚,这个反应具有很高的立体选择性,且收率高,这个反应得到的3-季碳氧化吲哚具有两个手性中心,ee值高达95%。反应如图10所示。
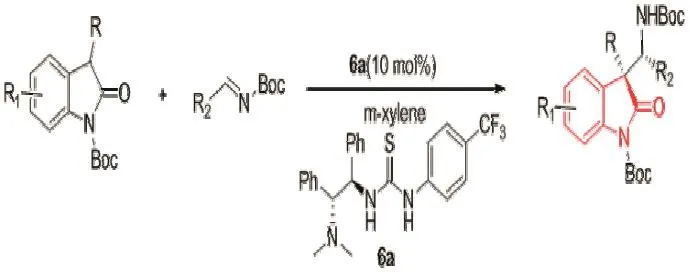
图10 通过mannich反应构建3-季碳氧化吲哚Fig.10 Construction of 3-quaternary carbon oxindole skeleton by mannich reactions
Liu Xiong-Li等[17]报道了绿色环保的合成3-季碳氧化吲哚的方法,特点是以水作为溶剂。3-取代氧化吲哚和甲醛水溶液和胺发生mannich反应,该反应在水中进行,SDS作为添加剂,反应如图11所示。在反应条件筛选中发现当使用多聚甲醛时,产率较低,使用福尔马林水溶液后,产率提高。有趣的是,在扩宽底物范围时发现,羟甲基化和mannich反应竞争,当使用开链二级胺时,发生mannich反应,当二级胺取代基团空间位阻较大或者是一级胺时,反应主要进行羟甲基化。

图11 通过mannich反应构建3-季碳氧化吲哚Fig.11 Construction of 3-quaternary carbon oxindole skeleton by Mannich reactions
1.6.通过aldol反应构建3-季碳氧化吲哚
自从脯氨酸被报道催化分子间直接羟醛反应以来,很多脯氨酸及其衍生物被应用到催化吲哚醌与丙酮等的不对称aldol反应。脯氨酸的衍生物主要是对羧基还原醇,接着可成醚,或者与胺发生酰化反应成酰胺等。2005年,Gianluigi Luppi等[18]报道了吲哚醌与丙酮在脯氨酸衍生物6b催化下发生aldol反应,因为脯氨酸衍生物溶解性不好,常需使用D MSO,DMF等大极性溶剂来增加溶解能力,且反应后处理不容易除去,但是当使用丙酮为溶剂时,丙酮既可作为溶剂又作为反应物,解决了这个问题。反应虽然产率非常高,几乎定量反应,但是对应选择性不是非常理想。反应如图12所示。
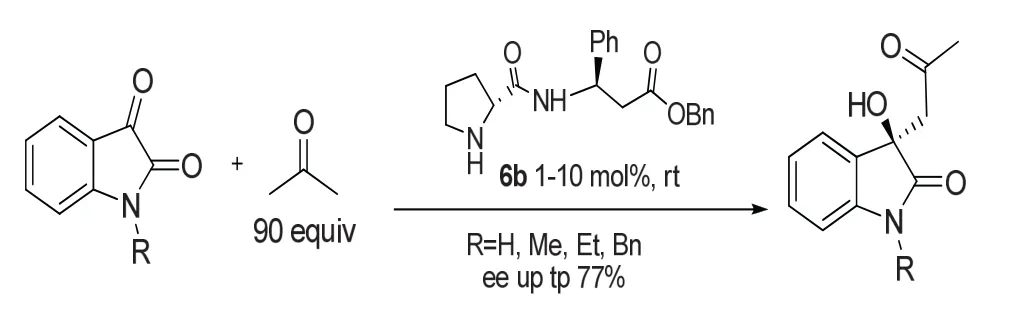
图12 通过aldol反应构建3-季碳氧化吲哚Fig.12 Construction of 3-quaternary carbon oxindole skeleton by aldol reactions
Chen Jia Rong等[19]合成了一类脯氨酸衍生物,催化吲哚醌与丙酮发生不对aldol反应,其中双功能催化的脯氨酸衍生物6c,催化该反应具有收率高,对映体过量值高的特点,反应如图13所示。在该反应条件下,不同取代基的吲哚醌均反应良好,产率很高(90%~99%),ee值77%~90%。但是当丙酮更换为2-丁酮时,反应会在两个位点,虽然总产率依然很高,但是目标化合物对映体过量值有所下降。

图13 通过aldol反应构建3-季碳氧化吲哚Fig.13 Construction of 3-quaternary carbon oxindole skeleton by aldol reactions
除了脯氨酸及其衍生物能够催化吲哚醌和酮的不对称aldol反应,一级胺和金鸡纳碱衍生物也能够催化该反应。Guo Qunsheng等[20]报道了金鸡纳碱的一级胺衍生物6d催化吲哚醌与乙醛发生不对称aldol反应,然后再硼氢化钠还原醛基,得到β-二醇氧化吲哚,该反应收率高,ee值最高达93%,当催化酮类化合物时,收率变化不大,ee值下降。反应如图14所示。
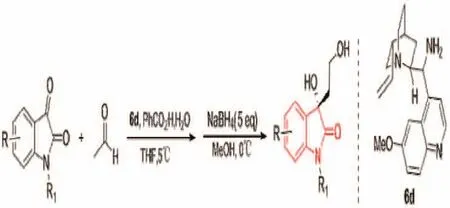
图14 通过aldol反应构建3-季碳氧化吲哚Fig.14 Construction of 3-quaternary carbon oxindole skeleton by aldol reactions
1.7.通过串联反应构建3-季碳氧化吲哚
天然产物的合成过程中,官能团保护、脱保护策略、中间产物分离提纯等问题一直困扰着研究者,而串联反应可克服以上诸多难题,串联反应具有无需分离反应中间体、产率高、选择性高、原子经济性等优点,新颖高效的串联反应在构建复杂螺环分子中起到了非常重要的作用,已被广泛地应用于有机合成研究中。Klaus Albertshofer等[21]报道了3-取代氧化吲哚与硝基烯在金鸡纳碱衍生物7a的催化下,发生不对称Michael-Henry串联反应,得到具有四个连续手性中心的螺环氧化吲哚。该反应收率高、非对映选择性好、ee值高。但是当氧化吲哚的苯环为吸电子基团取代时,产率和选择性稍有下降。通过3-取代氧化吲哚与硝基烯的Michael-Henry串联反应成功地构建了对人皮肤癌细胞有一定的活性的Cit rinadin B和Cyclopiamine B的结构骨架。反应如图15所示。
Artur Noole等[22]报道了3-烯基氧化吲哚与硝基酮在手性硫脲催化剂7b作用下,发生Michael-Aldol反应生成具有连续四个手性中心的螺环氧化吲哚化合物,经过反应条件筛选,当3-烯基氧化吲哚为1e q,硝基酮为2 eq,10 mol%7b,在二氯甲烷为溶剂,室温反应,该反应具有收率较高、dr值高、ee值高等特点,但是当硝基酮上取代基不同时,产率下降,ee值不变。该反应同样构建了活性分子Cyclopiamin e B的骨架结构,为这类化合物的合成奠定基础。反应如图16所示。
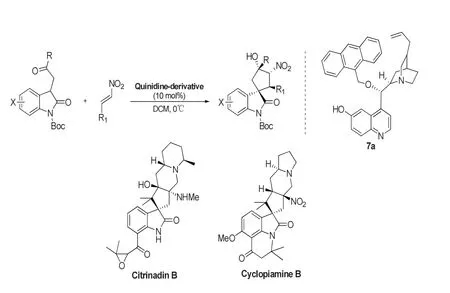
图15 通过串联反应构建3-季碳氧化吲哚Fig.15 Construction of 3-quaternary carbon oxindole skeleton by tandem reactions

图16 通过串联反应构建3-季碳氧化吲哚Fig.16 Construction of 3-quaternary carbon oxindole skeleton by tandem reactions
2.3-季碳氧化吲哚衍生物的抗肿瘤活性
3-季碳氧化吲哚衍生物是一类常见且重要的生物碱,3-季碳氧化吲哚骨架广泛地存在于天然活性化合物中,正是由于其特殊的骨架结构,决定了它具有广泛的生物活性,其中最为突出的是抗肿瘤活性,现在已被人们普遍关注,吸引了大量科研工作者,经过大量的研究,现在已经得到很多的先导化合物和一些临床药物。抗肿瘤药物主要通过以下几个生化机制来发挥其作用及功效:直接影响DNA的结构与功能;干扰核酸的生物合成;直接嵌入DNA干扰转录过程;干扰蛋白质的合成与功能;影响体内激素平衡;特异性酶及受体的抑制或阻断;诱导细胞分化[23]。
具有3-季碳氧化吲哚骨架的活性化合物如图17所示。SpirotryprostatinsA、B均是从肉汤发酵液里的曲霉素真菌里提取出来的,被发现是潜在的治疗癌症的药物[4];美国密西根大学王少萌课题组基于受体结构的设计和具有抗肿瘤活性SpirotryprostatinsA的特殊结构的启示,设计出一类新型的p53-MDM2结合抑制剂A-1,该抑制剂具有较好的p53-MDM2结合抑制活性[24],随后该课题组报道了MI-63具有p53-MDM2更好的结合抑制活性,在野生型p53的癌细胞里可以高效地与MDM2结合从而释放功能性的p53,激活了p53的功能,并可抑制肿瘤细胞的生长。而在p53缺失或敲除的癌细胞里,MI-63效果不显著,这个结果表明了MI-63对含野生型p53癌细胞具有高度选择性[25]。但是,MI-63的缺点是PK性质不理想,生物利用度不高,针对这些缺点,进行改造得到MI-147,且口服利用度也得到改善,并在SJSA-1异种移植模型,通过口服给药显著抑制了肿瘤的生长[26]。经过几年的努力,最终发现MI-73301,专利转让给赛诺菲公司,现在正在临床一期实验中[27-29]。Strychnofoline为包含有C3位季碳分子的双吲哚结构,可抑制许多细胞的有丝分裂,如在细胞mouse melanomaB16/Ehrich和Hepatom HW165中的有效抑制有丝分裂,对于黑色素瘤以及Ehrlich肿瘤细胞具有有效活性[1];化合物S1是孕酮受体的拮抗剂,在治疗不孕、子宫肌瘤、子宫内膜异位以及荷尔蒙引起的癌症方面有比较好的药效[30];Gelsebanine是从 Gelsemium Elegans(俗称“断肠草”)植物中分离出来的一种成分,具有抗肿瘤细胞活性[5]。A-2是Wang Shengzheng等[23]人发现的新型p53-MDM2结合抑制剂,具有较好的体外抗肿瘤活性,对A549、HCT116、MDA-MB-231这三个细胞株的IC50分别为1.67 μM、1.57 μM、3.55 μM,表现出与阳性对照药nutlin-3相当的抑制活性;化合物A-3是四川大学Zhang Li dan等[31]合成的新型螺环氧化吲哚衍生物,该化合物对A2870S和A2870T这两组肿瘤细胞具有较好的抑制作用;化合物A-4不仅具有非常好的体外抗肿瘤活性,对PLK-4肿瘤细胞的IC50为0.94 nM,还具有良好的体内抗肿瘤活性,对MDA-MB-468的裸鼠移植瘤具有96%的抑制率[32];A-5是一种具有良好肿瘤抑制活性的化合物,分别对MCF-7、PC-3、SKOV-3、U87、SMMC-7721、SY5Y、A875肿瘤细胞株具有较好的抑制活性,机制研究表明化合物是pim-1激酶抑制剂(IC50=5μM)[33]。
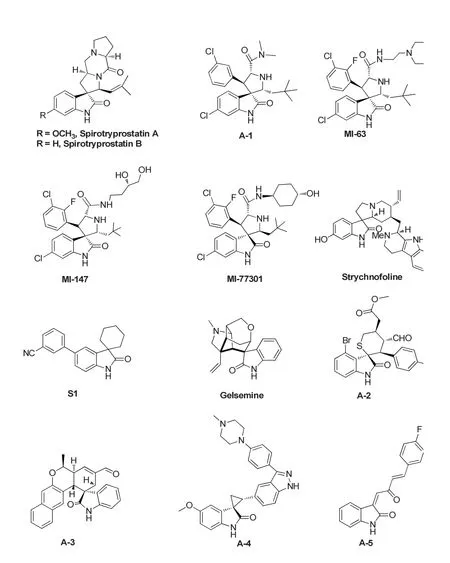
图17 具有3-季碳氧化吲哚骨架的活性化合物Fig.17 Active compounds with 3-quaternary carbon oxindole skeleton
3.结论及展望
3-季碳氧化吲哚类化合物自从被研究者们关注以来,大量的活性化合物被发现和合成,并有不少活性化合物正在进行药物临床前实验和药物临床实验,极有希望被开发成新药。所以发现3-季碳氧化吲哚类化合物的合成方法,不仅增加了新的3-季碳氧化吲哚骨架类型,还为全合成这类化合物提供了新的方法和思路;深度的筛选和发掘3-季碳氧化吲哚类化合物的生物活性,对新药的发现仍具有重要的意义。
参考文献:
[1]LERCHNER A,CARRERIRA E M.First Total Synthesis of(±)-Strychnofoline via a Highly Selective Ring-Expansion Reaction[J].Journal of the American Chemical Society,2002,124(50),14826-14827.
[2]WANG S M,SUN M,ZHAO Y J,et al.SAR405838:An Optimized Inhibitor of MDM2-p53 Interaction That Induces Complete and Durable Tumor Regression[J].Cancer research,2014,74(20):5855-5865.
[3]BADILLO J J,SILVA-GARIA A,SHUPE B H,et al.nantioselective Pictet-Spengler Reactions of Isatins for the Synthesis of Spiroindolones[J].Tetrahedron Letters,2012,43(6):5550-5553.
[4]AMMETTO I,GASPERI T,LORETO M A,et al.Chem In form Abstract: Synthesis of Functionalized Spiroaziridine-oxindoles from 3-Ylideneoxindoles:An Easy Route to 3-(Aminoalkyl)oxindoles[J].Chem in form,2010,41(15):6189-6197.
[5]XU Y K,YANG S P,LIAO S G,et al.Alkaloids from Gelsemium elegans[J].Journal of Natural Products,2006,69(9):1347-1350.
[6]MIYAKE F Y,YAKUSHIJIN K,HORNE D A.Preparation and synthetic applications of 2-halotryptamines:synthesis of elacomine and isoelacomine[J].Cheminform,2004,6(5):711-3.
[7]JIA Y X,KUNDIG E P.Oxindole synthesis by direct coupling of C(sp2)-H and C(sp3)-H centers.[J].Angewandte Chemie,2009,40(25):1636-1639.
[8]MOODY C L,FRANCKEVICIUS V,DROUHIN P,et al.Copper-catalysed approach to spirocyclic oxindoles via a direct C-H,Ar-H functionalisation[J].Tetrahedron Letters,2012,53(15):1897-1899.
[9]YUN L L,BO L W,JUN J C.et al.Organocatalytic Asymmetric Synthesis of Substituted 3-Hydroxy-2-oxindoles via Morita-Baylis-Hillman Reaction[J].Journalof the American ChemicalSociety,2010,132,15176-15178.
[10]CUI H L,PENG J,FENG X,et al.Dual Organocatalysis:Asymmetric Allylic-Allylic Alkylation of α,α -Dicyanoalkenes and Morita-Baylis-Hillman Carbonates[J].Chemistry-AEuropean Journal,2009,15(7):1574-1577.
[11]HE R,DING C,Maruoka K.Phosphonium salts as chiral phase-transfer catalysts:asymmetric Michael and Mannich reactions of 3-aryloxindoles[J].Angew Chem Int Ed 2009,48(25),4559-4561.
[12]DING M,ZHOU F,QIAN Z Q,et al,Organocatalytic Michael addition of unprotected 3-substituted oxindoles to nitroolefins[J].Organic&biomolecular chemistry,2010,8(13),2912-2914.
[13]LIU X W,YAO Z,YANG J,et al.1,3-Dipolar cycloaddition enabled isoxazole-fused spiropyrrolidine oxindoles syntheses from 3-methyl-4-nitro-5-alkenyl-isoxazoles and azomethine ylides[J].Tetrahedron,2016,72(10),1364-1374.
[14]DHANALAKSHMI P,BABU S S,HIMMARAYAPERUMAL S. One-pot chemo/regio/stereoselective generation of a library of functionalized spiro-oxindoles/ pyrrolizines/pyrrolidines from α-aroylidineketene dithioacetals[J].RSC Adv,2015,5(43),3705-33719.
[15]FILLATOV A S,KNYAZEV N A,MOLCHANOV A P,et al.Synthesis of Functionalized 3-Spiro[cyclopropa[a]pyrrolizine]-and 3-Spiro[3-azabicyclo[3.1.0]hexane]oxindoles from Cyclopropenes and Azomethine Ylides via[3+2]-Cycloaddition[J].Journal of OrganicChemistry,2017,82(2),959-975.
[16]XU T,KUN J.Organocatalytic Stereoselective Mannich Reaction of 3-Substituted Oxindoles[J].Organic letters,2008,10,3583-3586.
[17]LIU X L,ZHANG X M,YUAN W C,et al.A simple and eco-friendly method for the aminomethylation of 3-substituted oxindoles via three-component Mannich reaction in aqueous media[J].TetrahedronLetters,2011,52(8),903-906.
[18]GIANLUIGIL,PIER G C,MAGDAM,et al.Dipeptide-Catalyzed Asymmetric Aldol Condensation of Acetone with(N-Alkylated)Isatins[J].Journal of Organic Chemistry,2005,70(18),7418-7421.
[19]CHEN J R,LIU X P,ZHU X Y,et al.Organocatalytic asymmetric aldol reaction of ketones with isatins: straightforward stereoselective synthesis of 3-alkyl-3-hydroxyindolin-2-ones[J].Tetrahedron,2007,63(42),10437-10444.
[20]GUO Q,ZHAO J C.Primary amine catalyzed aldol reaction of isatins and acetaldehyde[J].Tetrahedron Letters,2012,53(14),1768-1771.
[21]KLAUSA, BIN T, CARLOS F, et al.Assembly of SpirooxindoleDerivatives Containing Four Consecutive Stereocenters viaOrganocatalytic Michael Henry Cascade Reactions[J]. OrganicLetters, 2012, 14(7), 1834-1837.
[22]ARTUR N, KAJA I, IVAR J, et al. Asymmetric Synthesis ofCongested Spiro-cyclopentaneoxindoles via an Organocatalytic CascadeReaction[J].Journal of Organic Chemistry,2013,78(16),8117-8122.
[23]尤启东.药物化学(第七版)[M].北京:人民卫生出012:258-293.
[24]DING K,LU Y P,NIKOLOVAKA-COLESKA Z,et al.Structure-based design of potentnon-peptide MDM2 inhibitors[J].Journal of the American Chemical Society,2005,127(29):10130-10131.
[25]DING K,LU Y P,NIKOLOVSKA COLESKA Z,et al.Structure-Based Design of Spiro-oxindolesas Potent,Specific Small-Molecule Inhibitors of the MDM2-p53 Interaction[J].Journal of Medicinal Chemistry,2006,49,3432-3435
[26]YU S,QIN D,SHANGARY S,et al.Potent and orally active small-molecule inhibitors of the MDM2-p53 interaction[J].Journal of medicinal chemistry,2009,52(24),7970-7973.
[27]ZHAO Y,LIU L,SUN W,etal.Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors[J].Journal of the American Chemical Society,2013,135(19),7223-34.
[28]SUN S H,ZHENG M,DING K,et al.A small molecule that disrupts Mdm2-p53 binding activates p53,induces apoptosis,and sensitizes lung cancer cells to chemotherapy[J].Cancer Biology&Therapy,2014,7(6),845-852.
[29]AGUILAR A,SUN W,LIU L,et al.Design of chemically stable,potent,and efficacious MDM2 inhibitors that exploit the retro-mannich ring-opening-cyclization reaction mechanism in spiro-oxindoles[J].Journal of medicinal chemistry,2014,57(24),10486-10498.
[30]JIANG K,JIA Z J,CHEN S,et al.Organocatalytic Tandem Reaction to Construct Six-Membered Spirocyclic Oxindoles with Multiple Chiral Centres through a Formal[2+2+2]Annulation[J].Chemistry,2010,16(9),2852-2856.
[31]ZHANG L, REN W, WANG X, et al. Discovery of novelpolycyclic spiro-fused carbocyclicoxindole-based anticanceragents[J]. European Journalof Medicinal Chemistry, 2017, 126,1071-1082
[32]LI SW, LIU Y, SAMPSON P B, et al. Design and optimization of(3-aryl-1H-indazol-6-yl)spiro[cyclopropane-1,3'-indolin]-2'-ones aspotent PLK4 inhibitors with oral antitumor efficacy[J]. Bioorganic&medicinal chemistry letters, 2016, 26 (19), 4625-4630.
[33]SUN H B,WANG X Y,LI G B,et al.Design,synthesis and biological evaluation of novel C3-functionalized oxindoles as potential Pim-1 kinase inhibitors[J].RSC Advances,2015,5(37),29456-29466.
猜你喜欢
中老年保健(2022年3期)2022-11-21
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
昆明医科大学学报(2020年12期)2021-01-26
陶瓷学报(2020年6期)2021-01-26
棉花学报(2020年3期)2020-08-08
人物画报(2020年36期)2020-03-13
科学与财富(2019年15期)2019-10-21
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
安徽医科大学学报(2015年9期)2015-12-16
