
植物药国际申报中毒理研究的关键问题
2018-05-02 06:38李旻,李华
中国药理学与毒理学杂志 2018年1期
李 旻,李 华
(国家上海新药安全评价研究中心,上海 201203)
中药作为中华民族的瑰宝,也是全世界人民共同的财富,绝不能固步自封,而应不断地发展和扬弃,中药国际化则成为其必然发展趋势。目前,我国的中药出口已经多达上百个国家和地区,其中亚洲地区既是中药出口的传统市场,也是主要市场,主要有日本、韩国、中国香港和东盟等区域。美国市场近几年也稳步增长,且大有跃居第一大市场的趋势,然而对于这些市场我国出口的主要是中药材、中药材提取物和保健品原料等附加值相对较低的产品,缺乏拥有自主知识产权的药品。欧盟是世界上最大的植物药市场,也是我国中药植物药出口的主要目标市场之一。欧盟市场,我国中药的出口情况与在美国市场的情况类似,且自《欧盟传统药注册程序指令》(Directive 2004/24/EC)于2011年4月30日废止后,我国中药出口欧盟就呈现下滑趋势。故本文将美国和欧盟植物药相关注册法规结合实例浅谈一些想法,以供同行参考和借鉴。
1 中国中药和欧美植物药的定义和分类
中国国家食品药品监督管理总局(China Food and Drug Administration,CFDA)认为,中药是指在我国传统医药理论指导下使用的药用物质及其制剂,并按是否在国内外上市、给药途径和剂型是否改变等特点分为9类。
美国食品药品监督管理局(Food and Drug Administration,FDA)认为,植物药涵盖植物材料、藻类、大型真菌及其组合产品,但不包括以下物质:①含有动物或动物器官(例如昆虫和环节动物)和(或)矿物,但当这些物质作为传统植物药制剂中的次要组分时除外(例如传统中药和印度草药);②源于植物物种的材料〔经过转基因技术(例如通过重组DNA技术或克隆)以期产生一个单分子实体〕;③通过酵母、细菌、植物细胞或其他微生物(包括作为底物的植物)发酵生成的产品〔如发酵工艺的目的是生成单分子实体(例如抗生素类、氨基酸类和维生素类)〕;④高度提纯原料药,源于天然来源(例如紫杉醇)或经化学修饰(例如合成自薯蓣提取物的雌激素类)。
欧洲药品管理局(European Medicines Agency,EMA)认为,植物药药品是指仅以一种或多种植物药物质、一种或多种植物药制剂以及一种或多种植物药物质与一种或多种植物药制剂复方作为活性组份的任何一种药用产品。其中植物药物质是指所有未经加工的植物全株、片段或切制的植物、植物部位、藻类、真菌和苔藓类,都可称为植物药物质,它们通常是干燥状态,但有时也是新鲜的。不经特殊处理的某些分泌物也可作为植物药物质;植物药制剂是指由植物药物质制备而得到,制备方法如萃取、蒸馏、压榨、分馏、纯化、浓缩和发酵。这些植物药制剂包括粉碎或粉状的植物药物质、酊剂、提取物、挥发油、压榨汁和经加工的分泌物等。
通过以上对比不难发现,中国的中药与欧美的植物药在定义和具体范畴上虽存在一些差异,但中国的中药基本可按照植物药的技术要求在欧美进行注册申请[1-3]。故以下统称为植物药。
2 美国和欧洲植物药申报现状
1999-2013年美国FDA共收到318个植物药新药研究申请(investigational new drug,IND)(图1),进入新药申请(new drug application,NDA)的更少。时至今日,美国FDA批准注册的植物药仅有2例,1例是2007年上市的绿茶提取物veregen(德国MEDI-Gene AG公司),仅作为外用药;另1例是2012年12月批准的第1例口服植物药Crofelemer(美国Salix制药有限公司)。让人遗憾的是,作为植物药传统强国,我国国内药企的植物药迄今仍无1例在美国注册上市成功,仅有个别植物药处于临床研究阶段。例如,2013年上海现代中医药股份公司的“扶正化瘀片”在美国完成Ⅱ期临床试验,2016年天士力制药集团股份有限公司的“复方丹参滴丸”在美国完成Ⅲ期临床试验。
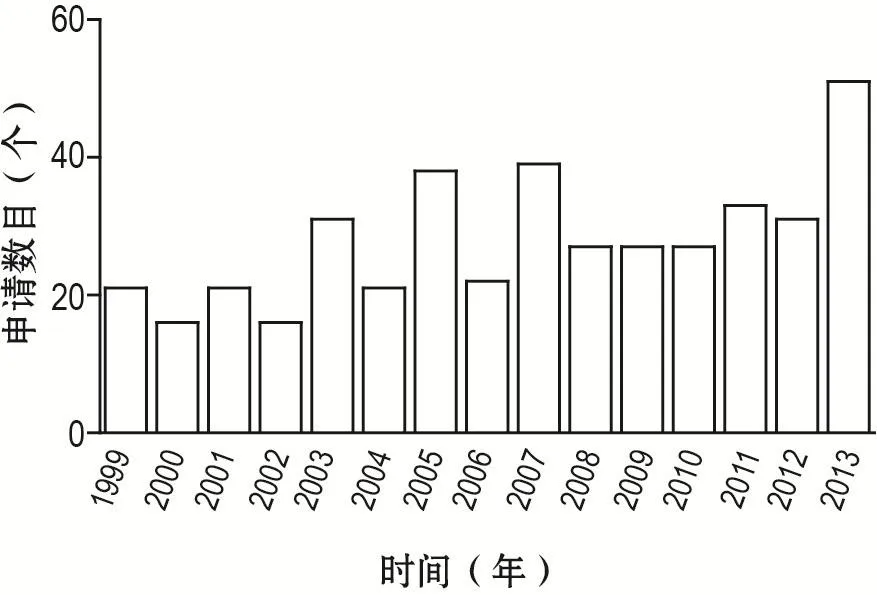
图1 1999-2013年美国食品药品监督管理局植物药新药研究申请数目[4].
欧洲植物药注册例数相对较多,2004-2015年共计1577个植物药获得在成员国上市许可(图2),而源自我国的植物药仅有2个品种注册成功,一个是地奥制药集团公司的地奥心血康胶囊(2012年3月,荷兰),另一个是天士力制药集团的丹参胶囊(2016年1月,荷兰)。

图2 欧洲植物药注册情况[5].
3 美国有关植物药注册申报要求中对毒理研究的规定
3.1 历史演变
美国FDA早在1996年8月16日开始起草与植物药相关的管理规定,在广泛征求意见,几经易稿后,于2000年8月在网上发布了《植物药研发工业指南(草案)》进一步征求意见,随后在2004年6月9日美国FDA正式发布了《植物药新药研究指南》(简称《指南》)。该《指南》对美国植物药市场产生举足轻重的作用,对世界各国植物药的相关管理法规也产生了不同程度的影响。在经历了11年的经验更替后,美国FDA药物评价和研究中心(CDER)于2015年8月17日发布了修订版的《植物药研发工业指南》,并在世界范围内征求意见,随后于2016年12月生效。
新《指南》(2015年版)特别对标题进行了改动,2004年版《指南》的标题为Guidance for Industry Botanical Drug Products,2015年新《指南》的标题改为Botanical Drug Development Guidance for Industry,重点从产品监管转向研发监管,突出了对植物药产品从研发到上市的全程监管。
新《指南》的编写遵照新药研发逻辑,在2004年版《指南》的基础上进行修改,内容和篇幅均有所精炼,但修改变动不大,保障了法规的连续性和一贯性,同时新《指南》和2004年《指南》中审评的基本理念和思路也无显著变化。新《指南》内容共分7个部分,包括简介、背景、一般管理办法、植物药研发的新药临床试验申请、Ⅰ期和Ⅱ期临床研究申请、Ⅲ期临床研究申请以及植物药新药上市申请。
3.2 对毒理学资料的要求
不同的临床研究阶段对毒理研究的要求不同。
3.2.1 Ⅰ期和Ⅱ期临床研究申请
用于支持植物药品Ⅰ期和Ⅱ期临床研究的非临床资料数量将取决于既往人体使用程度和所提出的临床研究设计,举例如下。
3.2.1.1 已在美国作为膳食补充剂合法上市销售的植物药
如拟定用途与既往使用类似,在无进一步非临床毒理学试验情况下,可允许开展初步的临床研究。然而应提供药品安全性相关的文献及其他现有资料。
3.2.1.2 目前未在美国合法上市销售的植物药
如用药途径、制备、加工和使用均符合范围广泛的既往人体使用经验,又无额外的非临床毒理学试验,上述信息如有充分的资料也可能足以支持初步临床研究。
3.2.1.3 临床研究中的预期用量超过既往人体用量
如剂量更高或持续时间更长,不论药品目前是否在美国合法上市销售,均应进行额外的非临床毒理学评价,保证充分解决既往人体使用与所提出的临床研究的差异。
3.2.1.4 植物药品的非传统用药途径
如传统为局部用药,但申请人拟用途径为口服,可能需要额外的毒理学资料,以便在初步临床研究前支持这种差异的安全性。
3.2.1.5 不具有广泛范围的既往人体使用经验的新植物药
需要进行详尽的非临床毒理学评价。这种评价类似于化学药的评价,建议参照相关的人用药品注册技术规定国际协调会议(International Confer⁃ence on Harmonization of Requirements for Regis⁃tration Pharmaceuticals for Human,ICH)和美国FDA指导原则。
3.2.1.6 未针对新药研究申请提交开展正式的非临床毒理学研究
申请人应开展文献检索工作,认定与下述安全性有关的公开可用资料:①拟采用商业化植物药的最终处方;②药品的植物药原料药;③植物药原料药已知的活性或化学成分。
上文所提及文献资料应依据《美国联邦法规》第21篇“食品与药品”中第312部“试验用新药申请”的第23(a)(8)(ii)节规定〔21 CFR § 312.23(a)(8)(ii)〕,提交来自于医学与毒理学文献库(例如Medline与Toxline)的现有数据的全面综述,以用于审评,并在初次IND申请中应解决以下问题:①一般毒性;②靶器官或系统毒性;③所有关于植物原料药各种成分的遗传或生殖毒性数据;④剂量与毒性反应持续时间之间的关系。
3.2.1.7 之前仅在美国以外国家或地区上市销售的植物药
申请人应提交经核实的既往人体使用安全性数据。
3.2.2 Ⅲ期临床研究申请
通常应提交来自于在动物中开展的标准毒理学研究的毒性数据,以支持后期临床研究。但是若改变了处方,则需要进行非临床桥接试验。申请人应与美国FDA新药办公室(Office of New Drugs,OND)审评部门就处方变更进行讨论,决定是否需要桥接或进行其他类型的研究以保证变更的合理性。
如植物药研发的任何阶段出现问题,鼓励申请人向美国FDA的OND审评部门咨询。在制定拟用于Ⅲ期临床研究的植物药非临床毒理学研发计划时,申请人应考虑下列要点。
3.2.2.1 安全药理学或毒理学
对于后期临床研究,植物药的安全药理学或毒理学评价与非植物药的相关评价并无差异。用于毒理学研究的剂量应符合ICH M3(R2)的原则。申请人应与相关的OND审评部门就安全药理学(参见ICH S7A和S7B)和毒理学研究达到可接受剂量水平而所计划采用的方法达成一致。原则上,毒理学研究中采用的最高剂量应产生某种程度上可评价的毒性。这些毒理学资料可为人体研究的安全性监测提供信息。
3.2.2.2 非临床药代或毒代动力学研究
由于植物药品通常含有一个以上的化学成分,使用标准的药动学方法在动物中证实全身暴露量,可能在技术上是一挑战。因而,采用灵敏的分析方法监测植物药品中具有代表性的化学成分,可提供全身暴露量相关的信息。如可行,应在药代或毒代动力学研究中评价对毒性造成影响的成分。申请人还应尽可能研究上述化学成分的代谢动力学参数。
3.2.2.3 生殖毒性
对于绝大部分的植物药品,在生殖安全性方面,与动物毒性研究相比,既往的人体数据可靠性较小。在缺乏经过确证的人体及动物生殖安全性数据情况下,生殖毒性研究通常应参照ICH指南(与非植物药的相同)开展。
3.2.2.4 遗传毒性研究
如之前未开展相关评价,应在Ⅲ期临床研究前开展全面的遗传毒性评价。遗传毒性标准组合的规定如下所示,具体参见ICH S2(R1)。
组合1:①细菌基因突变试验;②染色体损伤细胞遗传学试验(体外染色体畸变试验或体外微核试验)或体外小鼠淋巴瘤Tk基因突变试验;③体内遗传毒性试验(体内染色体畸变试验或微核试验)。
组合2:①细菌基因突变试验;②在2个不同组织的体内遗传毒性试验(通常是体内微核试验和非血液/骨髓组织的体内试验)。
对于植物药和非植物药而言,对上述标准组合检验结果解析以及是否需要开展额外试验的要求是一致的。
3.2.2.5 致癌研究
植物药品拟定使用的适应证和持续时间,将决定是否需要开展致癌性研究。应向FDA提交致癌试验方案,依照《特别方案评价》审查,在该试验启动前取得同意,以确保剂量选择和试验设计的合理性,且可被接受。
3.2.2.6 其他毒性研究
一般对于植物药而言,针对特定毒理学研究的建议(例如,用于确定潜在毒性的生物标志物或作用机制理解)与非植物药无区别。
3.2.3 植物药新药上市申请
考虑到植物药品的特性和注意事项,对于植物药品,新药申请前会议(the pre-NDA meeting)尤为重要。因为植物药新药申请的毒理学要求在很大程度上与非植物药一致,参照FDA相关指导原则。所以,申请人应与FDA就涉及研究用IND过程中所需的毒理学研究的所有监管事项紧密协作。建议应在Ⅱ期临床试验结束之前开始与FDA讨论在正常药品研发中可能的例外,在Ⅲ期临床试验结束之前达成总体一致,并根据“处方药申报者付费法案”(Prescription Drug User Fee Act,PDUFA V)[6]中绩效目标的规定,在新药申请前会议纪要中体现。
4 欧洲药品管理局植物药注册申报要求中对毒理研究的规定
4.1 历史演变
2001年颁布的《人用药品的欧共体法规2001/83/EC法令》,是欧盟管理人用药品的重要法规,其中对植物药并无特殊的政策。依照法律,植物药如果要在欧盟国家注册上市,必须与其他药品一样在安全性、有效性和质量方面按照相同标准进行评审。
基于欧洲有使用传统植物药的历史及其特殊性,1999年9月欧洲药品委员会成立了一个成员国工作组,研究建立一个可行的、适用于传统植物药品的法令。关于传统植物药品的法令草案经历了长达4年的讨论、谈判和修改,最终于2004年4月30日由欧洲议会和欧洲理事会通过。
2004年4月30日《欧盟官方杂志》用20种欧洲语言发表了欧盟关于传统植物药品的法令,即《2004年3月31日欧洲议会和理事会2004/24/EC法令,关于人用药品的欧共体法规2001/83/EC法令中传统植物药部分的修订》,对欧盟人用药品法令进行修改,主要添加了关于植物药的条例,并要求成员国在必要时应修改或制定本国的有关政策,以在2005年10月30日前符合该《法令》的规定。对于那些在该《法令》生效时,已经上市的传统植物药产品,该《法令》要求各成员国的主管部门在2011年4月30日前适用该《法令》。随后EMA结合法规2001/83/EC和2004/24/EC作为依据,于2006年出版了《植物药的非临床指导原则》[7]。与美国的情况相似,经过11年经验的更替,EMA于2017年8月出版了2006年版本的修订版草稿[8],该草稿主要对2006年版遗传毒性评价部分进行了修订,并增加了对辅料的关注度,其他原则基本未变。
4.2 对毒理学资料的要求
欧洲与美国的注册方式不同,《传统植物药注册程序指令》为仅限于在欧洲内有长期临床应用的植物药产品的简化程序,且这些植物药应符合如下标准:①适合于传统植物药产品的独特适应证,这些传统植物药产品的组成和用途,不需执业医师的诊断、处方或监督等干预下就能使用;②具有与特定作用强度和剂量相符的特定服用方法;③口服、外用和(或)吸入制剂;④已超过欧共体法规2004/24/EC法令第16c(1)(c)条规定的传统应用期,指在申请日之前已有至少30年的药用历史,包括在共同体内至少15年的使用历史;⑤有足够的药品传统应用资料,特别是被证明在特定条件下使用无不良反应的产品。
对于此类植物药通常基于悠久的人体使用经验,对于安全药理试验、单次给药毒性试验、重复给药毒性试验、免疫毒性试验和局部毒性试验,在申报时仅需提供安全性数据的文献综述和专家报告(不需要额外非临床毒理试验结果)。但应注意:①上述规定并不降低对安全性方面的要求,所提供的文献应全面覆盖欧共体法规2001/83/EC附件Ⅰ中的要求,且如有某方面数据的缺失应说明;②文献的来源和检索策略应说明;③对于不遵从现有法规如GLP的毒理学试验,应说明其数据可靠性的理由;④当不能排除对产品安全性的担心时,主管当局有权额外要求进行评价药品安全性的试验;⑤着重关注生殖毒性试验、遗传毒性试验和致癌试验。
4.3 对生殖毒性试验的要求
符合下述4个标准之一即可不用补充新的试验,否则在不明确、存疑或文献结果提示阳性的条件下应补充Ⅰ、Ⅱ和Ⅲ段生殖试验。①已有上市后安全性试验的结果或充分的流行病学调查结果;②系统性和综合性文献搜索结果的评估并结合上市后经验,未发现生殖毒性阳性结果,且妇女在怀孕或哺乳期间不会使用该药品;③已有怀孕妇女和新生儿的调查结果;④育龄妇女不会使用该药品。
4.4 遗传毒性试验
应评价植物药的遗传毒性。很多活性物质已可获得遗传毒性数据,但这些数据的质量常常不足以进行安全性评价。故当无充分的评价时,应进行遗传毒性试验,尤其应关注结果相应性和现有数据外推疑似阳性的植物药。
在可获得的遗传毒性资料不充足时,推荐先进行体外试验。在体外试验表现出阳性结果时,应进行恰当的研究(主要是体内试验)予以确证。按(图3)所示的策略进行。
4.5 致癌试验
结合植物药的用药周期,通常无致癌性的怀疑时,可不进行致癌试验;即使有致癌性的怀疑也可通过合理的评估排除致癌试验,应考虑以下几点:①致癌性的怀疑是否基于遗传试验,那么可在进一步的遗传试验(主要为体内试验)中是否可以确认或排除;②致癌性的怀疑是否基于一个可能的表观遗传学机制;③基于使用目的,已有的科学性数据(非临床、临床、流行病学调查和上市后)的程度和质量是否能充分地排除怀疑;④基于预期的获益,已有的科学性数据(非临床、临床、流行病学调查和上市后)的程度和质量是否能充分地证明获益大于风险。
4.6 毒代动力学研究
毒代动力学资料主要在新产品的动物实验中联合在一起要求。主要针对已知可被确定的具有药理活性一种或一组的中药组分和(或)具有特定毒性特征的一种或一组的植物药组分。
5 中美非临床药理毒理技术要求的差异
由于欧洲的要求相对于美国而言更为宽松,故本文主要比较中美之间的差异。

图3 遗传毒性分步评价步骤.
①美国FDA对于植物药无类别要求,均归属于植物药,与我国目前的分类管理有所不同。②美国FDA是分阶段申报,对于申请初期(Ⅰ和Ⅱ期)临床研究阶段,技术要求较为宽松。当申报扩大的临床(Ⅲ期)研究时,其技术要求基本等同于化学药,要提供遵循GLP规范的安全性评价试验。③美国FDA对植物药的毒理学研究要求较高,申报Ⅲ期临床时与化学药基本等同,但对药效学的要求较为宽松。我国对于毒理的要求则与申报的类别相联系,更注重药物的历史应用情况[9],且根据国家局相关文件精神,药品的有效性风险主要由申请人承担,所以目前国内对于药效学的要求也较宽松。④美国FDA对植物药申请初期临床试验、扩大临床试验和生产上市的非临床安全性研究要求中均未强制要求提交单次给药毒性试验资料。⑤加强对遗传毒性及生殖毒性研究的重视,我国的中药和天然药物在生殖毒性、遗传毒性以及致癌性研究方面的重视程度不如美国FDA对植物药的要求。⑥美国FDA对于植物药的前期人用经验对安全性的参考价值有一个相对合理的定位,他们主张先前的人用历史安全性参考信息仅支持植物药进入初期临床试验,而对于扩大的临床试验和批准上市申请必须有严格设计的、完整的非临床安全性研究资料予以支持。而我国往往过高估量了前期人用经验对中药和天然药物新药安全性的参考权重。⑦美国FDA强化GLP的实施执行,目前我国对于中药复方制剂的非临床安全性实验则仍未强制要求在通过GLP认证的实验室进行。⑧美国FDA对辅料非临床安全性研究非常重视,目前我国较缺失。
我国药监部门目前正处于深度改革中,管理理念和要求越来越国际化,笔者认为依据药物的风险进行分级管理且所有安全性试验应在GLP实验室进行将是大势所趋,故而目前中国与美国的差异很可能逐步缩小。
6 关注点
在植物药通往国际注册的道路上,对植物药安全性评价资料质量的重视程度应与化学药一致。①植物药的安全性评价应符合相应国家药监部门的要求。故而动态了解监管机构对植物药的看法十分重要,建议遵循“Case by Case”原则,申请人在收集与植物药有效性和安全性相关文献的同时,也应积极与审评部门进行有效沟通。②以临床试验和临床使用为终端目标,非临床安全性的研究和评价应立足于临床,应紧密结合临床适应证、目标人群、临床疗程和临床用药方法等情况以及与药监部门沟通的结果,考虑所需进行的非临床安全性评价试验范围,然后进一步细化每一个非临床安全性试验的设计和实施。③植物药相对于组分单一的化学药而言,影响质量的不可控因素更多,故而更应在具有良好质量管理体系的GLP实验室完成相应的安全性试验。④植物药组分复杂加之目前的技术手段有限,难以定量分析每一种组分,但是给药制剂质量的好坏从源头上决定了试验的质量,故而为检测其药效主要组分的稳定性、均一性、含量等化学特性,应及早完成供试品给药制剂分析方法学开发。这也为安全性试验的后续开展提供保障。⑤药物在体内的暴露情况,为科学合理地解释毒理学发现提供了重要的参考依据,故而应重视药效主要组分及其主要代谢物的毒代动力学研究。⑥在所有安全评价研究中,致癌性试验所占试验周期最为漫长,故而根据临床用药周期和植物药本身的特性,采用风险分析的方法,分阶段地合理开展致癌性研究对于缩短植物药的整个安评周期,降低研发成本意义重大。
7 结语
植物药在我国有悠久的应用历史,但由于监管理念和研发理念的差异,国内的许多中药企业和中药项目还有诸多差距。立足于国际监管理念,我们应改变传统思想,在重视植物药毒理学研究科学性的同时,更强调关注相应国家监管机构理念和技术要求。只有这样,我们才能更快更顺利地走出国门,尽早实现植物药的国际化,让植物药为世界范围内的患者作出更多贡献。
参考文献:
[1] CFDA.Drug Registration Regulations[EB/OL].(2007-07-10)[2017.11.02]http://www.sda.gov.cn/WS01/CL0053/24529.html
[2] FDA.Botanical drug development guidance for industry[EB/OL].(2016-12-27)[2017-11-02]https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm458484.pdf
[3] EMA.Directive 2004/27/ec of the european parlia⁃ment and of the council[EB/OL].( 2004-04-30)[2017.11.02]https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2004_27/dir_2004_27_en.pdf
[4] World Chinese Medicine News.The development status of international Chinese medicine[EB/OL](2016-06-05)[2017-11-02]http://www.worldtcm.org/160605/M05213448.shtml
[5] EMA.Uptake of the traditional-use registration⁃scheme and implementation of the provisions of Directive 2004/24/EC in European Union Member States[EB/OL].(2015-05-13)[2017-11-02].http://www.ema.europa.eu/docs/en_GB/document_library/Report/2011/05/WC500106706.pdf
[6] FDA.PDUFA reauthorization performance goals and procedures for fiscal years 2013 through 2017[EB/OL].(2012-01-17)[2017-11-02]http://www.fda.gov/downloads/forindustry/userfees/prescriptiondruguserfee/ucm270412.pdf.
[7] EMA.Guideline on non-clinical documentation for herbal medicinal products in applications for marketing authorisation(bibliographical and mixed applica⁃tions)and in applications for simplified registration[EB/OL].(2006-09-07)[2017-11-02]http://www.ema.europa.eu/docs/en_GB/document_library/Sci⁃entific_guideline/2009/09/WC500003576.pdf
[8] EMA.Guideline on non-clinical documentation in applications for marketing authorisation/registration of well-established and traditional herbal medicinal products [EB/OL].(2017-08-22)[2017-11-02]http://www.ema.europa.eu/docs/en_GB/document_li⁃brary/Scientific_guideline/2017/08/WC500233817.pdf
[9] Zhu FP.FDA and CFDA consideration for nonclinical studies and evaluation on pharmaco-toxicology of traditionalChinese medicine [J].TraditChin Drug Res Clin Pharmacol(中药新药与临床药理),2010,21(1):89-91.
猜你喜欢
当代水产(2022年6期)2022-06-29
华人时刊(2022年1期)2022-04-26
当代水产(2021年6期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年2期)2021-03-29
小哥白尼(野生动物)(2019年5期)2019-08-27
中国药理学与毒理学杂志(2017年5期)2017-01-15
中国药理学与毒理学杂志(2017年4期)2017-01-15
创业家(2015年9期)2015-02-27
创业家(2015年9期)2015-02-27
