
某地下水有机污染场地污染源识别及自然降解研究
2018-04-04 08:33赵龙
城市地质 2018年1期
赵 龙
(北京市地质环境监测总站,北京 100195)
0 前言
许多有机化工厂排放的有机污染物具有致畸、致癌、致突变的特性。近年来由于疏解非首都功能区,造成有机化工厂搬迁,其不合理废物排放造成的水土污染已成为重要的环境问题(高存荣等,2011;郭琳,2013),尤其是当污染物进入地下水时,其污染范围必然扩大,污染物也可能更难以降解。当前如何快速、经济、有效地治理有机污染,降低地下水环境风险是国内外学者共同关注的问题(Majone et al,2014;Kao,2014)。自然衰减法(MNA,Monitored Natural Attenuation)由于对环境干扰小、修复费用低、不产生次生污染而广泛应用于有机污染地下水修复中(Paradis,2014;Agah etal,2013)。该技术实施前需进行场地自然降解能力评价,主要确定能否进行自然降解及了解自然衰减的机理(何江涛等,2004)。因此评价中首要问题是了解污染场地的污染源类型及分布状况,而后对污染源进行自然衰减评价,为后期修复工作提供决策支持。
本研究针对某化工厂排污渗坑渗漏,以及其它可能因素引起的浅层地下水污染进行了调查,对污染物来源进行了综合分析,并对其自然降解情况进行了评价,研究成果可为我国地下水有机污染场地的调查与评价提供借鉴。
1 研究区概况
研究区位于河北沧州境内,区域属暖温带半湿润大陆性季风气候区,多年平均降雨量为552.6 mm,多年平均蒸发量为1252.4 mm,强烈的蒸发导致产生区域大面积盐碱地。场地面积约4 km2,属华北古黄河冲积平原,地势总体上南高北低,地面局部高差较小(高程起伏在50 cm左右)。
地下水主要赋存于沉积厚度为350m左右的第四系松散地层中,为多层结构的含水岩系。运用直推钻进技术(direct push technology,DPT)揭露了距地表25 m范围内的岩性变化情况,从上至下依次为粉土、粉质粘土、粉砂,部分层位夹层现象明显(图1、图2),粉质粘土与粉土相互交错沉积使得研究区单元较为封闭,受到黏土层的天然屏障作用,外界污染物很难进入地下水造成污染。研究区地下水水位在2m左右,主要补给源为降雨及河流侧向补给,以蒸发、侧向径流及人工开采为主要排泄方式。为满足调查目的,2012年在渗坑北侧1km范围内建立7处监测井,并在2012、2013年进行两期取样监测,同时结合民井取样数据对典型剖面的污染范围及自然衰减特性进行分析。
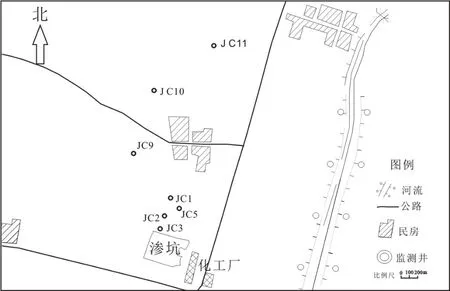
图1 监测孔分布Fig.1 The distribution of monitoring well

Fig.2 typical borehole pro file
2 污染物时空分布
2012年1月、2013年4月两次取样的实验室全扫描结果显示:研究区主要污染物包括单环芳香烃(Monocyclic Aromatics hydrocarbon,MAHs)、氯代烃(chlorinated hydrocarbons,CHCs)、及邻苯二甲酸酯类( Phthalate Esters,PAES)。MAHs总体浓度变化在0~32.5μg/L之间,其中井号为JC9的 MAHs浓度最高为32.5μg/L 。MAHs各组分中仅甲苯在各监测井中检出,浓度位于3.4~18.3μg/L之间,其余苯系物只在部分井中检出,浓度较低。总体上MAHs各组分含量均低于地下水质量控制Ⅱ级标准。CHCs浓度变化位于31.2~851.3μg/L之间,以《地下水质量标准》中Ⅲ类水为基准,计算发现CHCs中1,2二氯乙烷和氯苯均超标,超标率分别为100%和6.90%。其中1,2二氯乙烷浓度变化位于29.5~825μg/L之间,均值为315μg/L;氯苯浓度变化位于0.6~785μg/L之间,均值为123.12μg/L。依据地下水质量控制标准,氯苯浓度达到水质Ⅲ级标准,而1,2二氯乙烷均值已超过地下水Ⅴ级标准上限8倍,可见该区域受到严重的CHCs污染。
2.1 时间动态变化特征
对比两次分析结果(图3)可以看出,地下水中MAHs中整体浓度呈衰减趋势,变化明显,呈现出区域不均匀性,除JC9井点变化异常外其余井点变化在3.4~17.3μg/L之间。相比于MAHs,CHCs衰减程度更为明显,除JC11井点异常外其余井点地下水中CHCs浓度变化介于183~595.3μg/L范围内。研究区地下水中MAHs及CHCs总体上明显呈下降趋势,由于监测期是冬天,场地又位于耕地内,可基本排除外界的影响,所以两次监测结果可以初步反映研究区污染物的自然衰减过程。

图3 MAHs时空变化(左)CHCs时空变化(右)Fig.3 Temporal and spatial variation of MAHs(left),CHCs(right)
2.2 水平分布
沿JC3—JC11剖面,随着距离的增大,地下水中MAHs浓度变化波动较大,JC9井中MAHs浓度达到最高值32.5μg/L,随后浓度开始下降,到达JC11时浓度变为24.1μg/L。而地下水中CHCs浓度随着距离的增大,呈现先下降又上升趋势,最高浓度位于JC11井口830.6μg/L(图3)。
3 场地污染源识别
3.1 污染物主成分分析
对2012年12月及2013年4月研究区域地下水分析数据进行整理、分析,结合地下水风险指数(GUS)。GUS是由Gustafson(2010)提出,用来描述化学物质迁移性的经验性公式,该公式涉及土壤有机碳吸附系数、半衰期两种参数(王昭等,2009)。将GUS值(淋溶迁移值)较低的组分筛除,得到15种有机污染物数据。利用SPSS软件中因子分析程序对15种因子进行主成分分析。分析结果见表1,可以看出通过因子分析将原有数据值筛选出三个主成分因子,特征值分别为4.37、2.166、1.769,最大方差贡献率为43.7%,累积方差贡献率为83%,说明3个因子可以反应总污染样本的83%的信息。
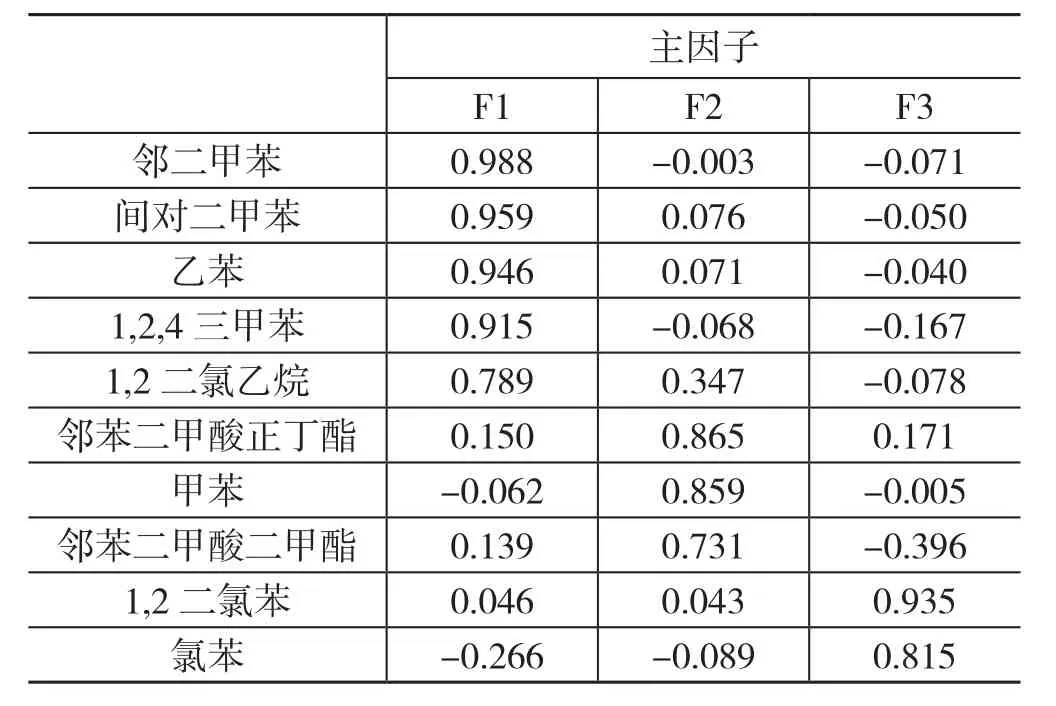
表1 主成分因子分析结果Tab.1 The result of principial component factor analysis
第一主因子F1表明,污染主要受到二甲苯(邻、间、对)、乙苯及1,2,4三甲苯(下文仅用英文字母MAH1表示)。因子载荷矩阵显示三者相关程度较高,推测三者来自同一个污染源,且污染特性相似。
第二主因子F2的贡献率为21.67%,其中甲苯、邻苯二甲酸酯类的因子载荷较大,与F2呈正相关。其中甲苯及邻苯二甲酸正丁酯相关性一般(相关系数为0.559),而二者与邻苯二甲酸二甲酯相关性较弱。
第三主因子F3显示,1,2二氯苯、氯苯因子载荷较大,与F3呈正相关,表明研究区受到1,2二氯苯、氯苯共同作用影响。
3.2 污染源辨识
依据室内分析结果,二甲苯、乙苯及1,2,4三甲苯仅在井点JC5、JC9、JC11处检出,浓度呈现随机性变化。研究表明,二甲苯、乙苯均为汽油和煤焦油的副产品,而1,2,4三甲苯为汽油、药物的成分之一。因此推测污染源可能来自地表含油物质经过降雨淋溶作用渗入地下水,或为成井过程中,通过钻机直接带入监测井内的外源污染物。
F1中除MAHs1外1,2-二氯乙烷也对区域污染具有一定贡献,通过对比距离渗坑最近的JC3号井点及距离渗坑最远的JC11号井点发现,JC11号井点中的1,2二氯乙烷浓度偏高,相比于JC3中1,2二氯乙烷浓度整整高出11倍。1,2-二氯乙烷主要用作工业生产四氯乙烯、氯乙烯及其它制剂的合成中间体,也用作酯类、橡胶、油漆、磷、碘等的溶剂。同时1, 2-二氯乙烷还被广泛地用作杀虫熏蒸剂,灭杀农作物和土壤中的虫害(孔祥斌等,2013;张大定等,2012)。对采样点1,2二氯乙烷检出浓度进行等值线绘制,发现1,2-二氯乙烷高值点均位于农耕地,监测区内JC3、JC1、JC10位于农耕地中边缘,三者1,2-二氯乙烷浓度较低为75、262、247μg/L。从图3中可以看出2012年12月份至2013年4月份期间(未耕作期),1,2二氯乙烷浓度急剧下降。位于JC3井点内浓度变化很小,其余浓度均有大幅度变化,其中JC11由原来825μg/L降到234μg/L降幅接近4倍。因此1,2-二氯乙烷受人为耕作影响较大,因此可判断1,2-二氯乙烷污染源可能来自杀虫剂的使用,与渗坑污染源污染无关。
因子分析结果显示F2包含甲苯、邻苯二甲酸酯类两种特征污染物,研究表明邻苯二甲酸正丁酯作为塑料增塑剂而被广泛使用(肖乃玉,2010),相比之下邻苯二甲酸二甲酯长用于化工涂料生产(王春等,2007)。因此前者广泛存在于环境中,已成为地表水、土壤中普遍检出的污染物。利用绘图软件绘制二者浓度等值线发现,邻苯二甲酸二甲酯与邻苯二甲酸正丁酯等值线不同,图中显示邻苯二甲酸二甲酯由渗坑向东北方向浓度逐级递增,到东北方向河道旁井中浓度为最大值560μg/L。而邻苯二甲酸正丁酯呈现出点状扩散污染,且农田中该物质含量较多。分析认为取样测试的水井位于棉花地中,当地农民常在棉花地上铺一层塑料地膜,地膜经过长期的降雨冲刷及地表径流,使地膜上的有机物(增塑剂)通过土壤孔隙运移污染区域地下水(肖乃玉等,2010;张英等,2011)。
区域甲苯浓度低,但覆盖面广,从上游到下游均有不同程度污染。从剖面甲苯浓度时空分布来看,监测区间内甲苯浓度显著下降,最大降幅达17.3μg/L,空间分布上,浓度变化无规律性,总体上分布均匀。因此F2可识别为面源污染源,与渗坑无关。
根据上述结果,考虑到污染物1,2-二氯乙烷并非来源于渗坑故将其剔除。剔除后污染物时空分布图(图4)可以看出CHCs最高值出现在距离渗坑最近的井点JC3,其余各点CHCs随着距离的增加逐渐下降,至JC11浓度变为1.3μg/L。对厂区生产工序调查中发现,厂区生产碱性红及OB酸的原料中包括氯苯及二氯苯,同时渗坑水样检验结果显示,检出高剂量的氯苯及二氯苯,其中氯苯浓度可达1280μg/L。据此可以佐证氯代烃为渗坑污染物,同时对区域地下水污染具有贡献作用。第二次测试值显示距离渗坑最近的JC3井中氯苯浓度有显著上升,由413μg/L升至785μg/L,可看出渗坑仍继续作为污染源污染着周围的地下水。F3为来源自渗坑的污染。

图4 剔除1,2二氯乙烷后CHCs浓度变化(左);甲苯浓度时空变化(右)Fig.4 The concentration of CHCs after remove 1,2 dichloroethane(left);Temporal and spatial of toluene(right)
4 典型污染组分氧化还原特征
以上分析结果表明,研究区起主要贡献作用的为氯代烃、甲苯污染,通过对比污染物浓度时空变化发现,选定剖面上污染物浓度有降低趋势。氧化还原电位(ORP)及溶解氧(DO)监测结果表明地下水主要为厌氧环境,因此考虑在此背景下污染物浓度下降与地下水氧化还原特征具有一定关系。结合室内无机分析结果,以位于监测剖面西北侧民井作为背景点,对区域主要电子受体进行分析。
厌氧条件下,硝酸根(NO3-)首先作为最终电子受体被消耗(Rifai,1995)。剖面水样检测结果显示,NO3-除JC9检出外,其余浓度均低于检出限,通过对比背景点水样发现,研究区总体NO3-浓度均在检出限附近波动,如此低浓度的NO3-不能作为电子受体被消耗,因此对区域氧化还原贡献较小。地下水中Fe2+浓度含量为0~0.41mg/L,但Mn2+浓度较高,剖面内平均浓度达 2923 mg/L,并且沿地下水流向Mn2+浓度有增大趋势,最高值点位于JC11处,浓度达4650 mg/L,超出背景值26倍之多(图5)。剖面上离子具有减小趋势。同时浓度与浓度具有反向相关性,依据还原反应式,反应中可产生,影响地下水的pH值(路莹,2013),另外在氧化还原作用下,有机碳源会被分解成无机碳源,最终以CO2形式排放,部分CO2溶于水以形式存在。监测的背景点浓度为518mg/L,低于剖面检出浓度最低值。因此可以判定污染物可能通过还原发生降解反应。

图5 典型电子受体区域分布Fig.5 Spatial of typical electron acceptor

图6 典型电子受体与CHCs之间关系Fig.6 The relationship between typical electron acceptor and CHCs
研究区内地下水pH在6.78~7.13之间,在该范围内MAHs类物质氧化还原电位值为300 mv(Wiedemeier,1996),而地下水ORP值位于-18.1~-110.2mv区间内,不满足MAHs类物质发生氧化还原自然降解,因此推测MAHs类物质可能由于挥发或其它物理过程降低浓度(Barker,1987;Nobre,2004;周睿等,2009)。Kao等在场地原位自然生物降解试验中发现,在土著微生物催化作用下CHCs可进行还原脱氯作用发生降解,在空间分布上地下水中ORP的值从-96 mv变化到-55 mv,同时在监测使用人工湿地对地下水中氯苯类污染物的移除中得出了类似的结果(Braeckevelt,2001),发现在此还原环境中可作为电子受体参与氯苯类还原降解反应。本研究区内ORP位于-18.1~-110.2mv之间,该电位符合CHCs还原降解条件(Wiedemeier,1996;Barker,1987;Nobre,2004),通过分析CHCs与电子受体(铁、硫酸根)之间的关系(图6),发现剖面上CHCs浓度变化与电子受体变化具有一定相关性,这与Schmidt(2014)、Bouwer(1984)等描述的污染物自然衰减中电子受体与污染物浓度变化关系一致,因此推测该区域可能发生CHCs还原降解。研究区内CHCs的生物还原降解过程,有待后续室内生物试验加以验证。
5 结论
对研究区地下水进行取样分析,结果显示区域地下水受到由多种污染物贡献的混合型有机污染影响,包括MAHs、CHCs及邻苯二甲酸酯类。
结合因子分析法将区域污染物源分为3类,石油污染源、环境面源污染及渗源污染。结合研究区内无机离子中典型电子受体的氧化还原特性,认为本区域发生了Mn、Fe和SO42-的还原降解反应,氧化还原电位较低。依据两期监测结果发现,在研究区地下水环境条件下,其氧化还原电位有利于CHCs类物质发生还原生物降解作用,且区域内电子受体浓度变化与CHCs浓度具有一定相关性。因此推测研究区可能发生了CHCs类污染物自然衰减,由于区域Fe3+浓度较低,未能充分参与反应,导致降解速率变慢,后期可通过加入Fe3+溶液类电子受体,提高反应速率,或使用化学试剂降低地下水Eh值,使污染物充分降解。
统计分析法可作为初步自然衰减评价的有效手段,对污染源类型,贡献及特征进行分析了解,结合电子受体分析,可更好地诠释地下水环境下的氧化还原特征,为后期进行地下水治理及修复提供有效的基础资料。
高存荣,王俊桃,2011.我国69个城市地下水有机污染特征研究[J].地球学报,(5):581-591
郭琳,2013.上海某污染场地浅层地下水中氯代烃自然降解机制及能力研究[J].环境科技,(3):9-13.
Majone M, Verdini R, Aulenta F, et al,2014.In Situ Groundwater and Sediment Bioremediation: Barriers and Perspectives at European Contaminated Sites[J].New biotechnology.
Kao C M,2014.Recovery of Chlorinated Solvent Trichloroethylene Contaminated Groundwater Using a Hybrid Treatment System[J].International Journal of Environmental Science and Technology, 11(1): 149-158.
Paradis C J,2014.Characterization of Anaerobic Natural Attenuation of Petroleum Hydrocarbons[D].UNIVERSITY OF CALIFORNIA, DAVIS.
Agah A, Ardejani F D, Ghoreishi H,2013.Two-Dimensional Numerical Finite Volume Modeling of Processes Controlling Distribution and Natural Attenuation of BTX in the Saturated Zone of a Simulated Semi-Confined Aquifer[J].Arabian Journal of Geosciences, 6(6):1933-1944.
何江涛,史敬华,崔卫华,等,2004.浅层地下水氯代烃污染天然生物降解的判别依据[J].地球科学,(3):357-362.
王昭,石建省,张兆吉,等,2009.华北平原地下水中有机物淋溶迁移性及其污染风险评价[J].水利学报,(7):830-837.
Gustafson D I,2010.Groundwater Ubiquity Score: A Simple Method for Assessing Pesticide Leachability [J] .Environmental Toxicology and Chemistry, 8 (4) :339-357.
王春,李晓东,史玉坤,等,2007.南通市地表水中邻苯二甲酸酯类污染状况研究[J].南通大学学报(医学版),(3):167-170.
肖乃玉,陆杏春,郭清兵,等,2010.塑料食品包装中邻苯二甲酸酯类增塑剂迁移研究进展[J].包装工程,(11):123-127.
张英,孙继朝,陈玺,等,2011.东莞地下水邻苯二甲酸酯分布特征及来源探讨[J].环境污染与防治,(8):57-61.
Rifai H S, Newell C J, Miller R N, et al,1995.Simulation of Natural Attenuation with Multiple Electron Acceptors[M].R.Hinchee, J.Wilson, and D.Downey, Battelle Press, Columbus, Ohio.
路莹,2013.浅层地下水系统石油类污染物的生物降解机制研究[D].吉林大学.
Verardo E, Saada A, Blanc C, et al, 2013.A Case Study of Monitored Natural Attenuation of Petroleum-Hydrocarbon Contaminated Soil[J].AquaConSoil 2013 Proceedings.
Corseuil H X, Monier A L, Fernandes M, et al, 2011.BTEX Plume Dynamics Following an Ethanol Blend Release: Geochemical Footprint and Thermodynamic Constraints on Natural Attenuation[J].Environmental science & technology, 45(8):3422-3429.
Wiedemeier T H, Wilson J T, Hansen J E, et al, 1996.Technical Protocol for Evaluating Natural Attenuation of Chlorinated Solvents in Groundwater.Revision 1[R].AIR FORCE CENTER FOR ENVIRONMENTAL EXCELLENCE BROOKS AFB TX.
Barker J P, Patrick G C, Major D, 1987.Natural Attenuation of Aromatic Hydrocarbons In a Shallow Sand Aquifer[J].Groundwater Monitoring & Remediation, 7(1): 64-71.
Nobre R, Nobre M M,2004.Natural Attenuation of Chlorinated Organics In a Shallow Sand Aquifer[J].Journal of Hazardous Materials, 110(1): 129-137.
Kao C M, Prosser J, 1999.Intrinsic Bioremediation of Trichloroethylene and Chlorobenzene: Field and Laboratory Studies[J].Journal of Hazardous Materials, 69(1): 67-79.
Braeckevelt M, Reiche N, Trapp S, et al, 2011.Chlorobenzene Removal Efficiencies and Removal Processes in a Pilot-Scale Constructed Wetland Treating Contaminated Groundwater[J].Ecological Engineering, 37(6): 903-913.
周睿,赵勇胜,任何军,等,2009. BTEX在地下环境中的自然衰减[J].环境科学,(9):2804-2808.
Schmidt M, Wolfram D, Birkigt J, et al, 2014.Iron Oxides Stimulate Microbial Monochlorobenzene in Situ Transformation in Constructed Wetlands and Laboratory Systems[J].Science of The Total Environment, 472: 185-193.
Bouwer E J, McCarty P L, 1984.Modeling of Trace Organics Biotransformation in the Subsurface[J].Ground Water, 22(4):433-440.
孔祥斌,2013.饱和含水层中1,2-二氯乙烷运移模拟及生物降解研究[D].河北农业大学.
张大定,谢云峰,柳晓娟,等,2012.1,2-二氯乙烷自然衰减过程中模拟饱和含水层的氧化还原条件变化[J].环境科学研究,25(12):1398-1403.
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
环境卫生工程(2021年1期)2021-03-19
聚氯乙烯(2018年2期)2018-07-05
上海农业学报(2017年3期)2017-04-10
化工管理(2017年9期)2017-04-10
中国药理学通报(2014年2期)2014-05-09
同位素(2014年2期)2014-04-16
浙江科技学院学报(2014年6期)2014-02-28
无机化学学报(2014年3期)2014-02-28
中国氯碱(2014年10期)2014-02-28
