
毛细管电色谱-激发诱导荧光检测动物性食品中多肽类抗生素
2018-04-02 05:23雷霄云宋云萍李茜诺张冰宇吴晓苹福州大学化学学院食品安全与生物分析教育部重点实验室福建福州350108
色谱 2018年3期
雷霄云, 宋云萍, 李茜诺, 张冰宇, 吴晓苹(福州大学化学学院, 食品安全与生物分析教育部重点实验室, 福建 福州 350108)
多肽类抗生素(peptide antibiotics, PTs)是衍生自氨基酸的一类抗生素的总称,主要作为药物饲料添加剂以促进动物生长及治疗畜禽疾病。随着动物养殖生产对兽药饲料依赖程度的提高,多肽类抗生素的安全性引起了人们的广泛关注。研究表明,PTs易残留于动物体内,并通过动物源性食品在人体富集,对肾、神经系统产生一定的毒性[1,2]。兽用PTs均为环状肽类,欧盟、日本、美国、澳大利亚等均规定了牛奶中多种PTs的限量值[3],主要包括杆菌肽A(100~400 μg/kg)、粘杆菌素(10~50 μg/kg)及维吉尼霉素(10 μg/kg)等;我国也规定了牛奶中杆菌肽A和粘杆菌素的限量值(500和50 μg/kg)。
常用于PTs快速筛检的分析方法有微生物法[4]、免疫分析法[5]等,但其选择性和定量能力相对较低。高效液相色谱法(HPLC)[6-10]是兽用PTs主要的分离分析方法,由于多数PTs结构中缺乏发色团,相关组分众多,甚至在一个分子中含有同一氨基酸的两种构型[11],导致分离困难,且UV检测器的灵敏度无法满足PTs残留限量的检测需求,一般需要使用造价昂贵的质谱检测仪[12]。我国国家标准方法[13]采用梯度洗脱的LC-MS/MS法测定牛奶、奶粉或猪肉、猪肝和猪肾中的杆菌肽以及饲料中的硫酸粘杆菌素[14],但是方法的预处理操作繁琐、使用成本高,限制了其实际生产应用。因此发展简便、选择性和灵敏度高、成本低的多肽类抗生素分析新技术十分必要。
毛细管电色谱兼具高效液相色谱和毛细管电泳的双重分离机制[15],对中性物质和带电物质均可分离,尤其是结合灵敏度与选择性极强的激光诱导荧光(LIF)检测技术,检出限可达到10-15g甚至10-18g水平,对于样品中痕量甚至超痕量残留药物的分析具有很好的应用前景[16]。LIF检测中,针对目标物建立的高效衍生方法是决定方法选择性和灵敏度的主要因素。然而,目前尚缺乏有关多肽类抗生素荧光衍生方法的研究。苯并呋咱类荧光衍生试剂克服了一般衍生反应耗时、衍生副产物多、衍生效率低等缺点,为痕量或超痕量物质标记提供了新方向。4-氟-7-硝基-2,1,3-苯并呋咱(NBD-F)是一种与伯胺、仲胺反应速度较快的高效苯并呋咱类衍生试剂[17],常用于氨基酸和小分子胺的衍生和检测[18]。Aoyama等[19]以NBD-F为柱前衍生试剂,结合HPLC-荧光检测(FD)分析了大鼠血浆中的氨基酸,检出限可达2.8~20 fmol; Zhang等[20]采用NBD-F荧光衍生结合毛细管区带电泳(CZE)-LIF联用技术,快速灵敏地分离检测了食品中的精胺和组胺;Carlucci等[21]以NBD-F荧光衍生结合HPLC-二极管阵列检测(PDA)-FD联用技术检测人胰液样品中奥曲肽和加贝酯等药物的含量,检出限为0.025~0.05 μg/mL; Wu等[22]在聚合物整体柱上采用LC-FD方法,在6 min左右分离了精氨酸等氨基酸对映体的NBD-F荧光衍生产物。但是目前尚未见有关NBD-F衍生化多肽类抗生素的报道。
本文将CEC-LIF联用技术应用于多肽类抗生素的分析,以NBD-F为柱前荧光衍生试剂,发展了多肽类抗生素高效的衍生方法。通过优化影响电色谱分离与LIF检测的实验条件,建立了环状多肽类抗生素的CEC-LIF分析方法,并将其应用于牛奶和饲料样品的检测,方法简单,样品和溶剂耗量小,成本低,灵敏度满足我国对多肽类兽药的残留检测要求。
1 实验部分
1.1 仪器、试剂与材料
自组装毛细管电色谱分析系统[23],包括TriSep-2010型微流控系统、±30 kV高压电源(上海通微分析技术有限公司)、LC-10 ADvp高压液相色谱泵(日本岛津公司)和半导体泵浦式激光诱导荧光检测器(中国科学院大连化学物理研究所); Eclipse荧光分光光度计(美国瓦里安公司); HB-031金属恒温加热器(上海申能博彩生物科技有限公司); WH-2微型漩涡混合仪(上海沪西分析仪器厂有限公司); Oasis HLB固相萃取柱(500 mg/6 mL,美国Waters公司); 0.22 μm微孔滤膜(上海市新亚净化器件厂)。
杆菌肽(BAT, 60 U/mg,比利时Acros Organics公司)、硫酸多粘菌素B(PLM-B, 6 000 U/mg,美国Sigma公司)、硫酸粘杆菌素(CLS, 15 000 U/mg,美国Sigma公司)。NBD-F(纯度99%,日本东京化成工业株式会社);磷酸二氢钾(KH2PO4)、磷酸氢二钾(K2HPO453H2O)(分析纯,中国(上海)化学试剂研究所);乙腈和甲醇(色谱纯,上海国药集团化学试剂有限公司)。其他试剂均为分析纯;分析用水为Milli-Q(美国Millipore公司)制备的超纯水(18.2 MΩ5cm)。牛奶和饲料样品分别购自福州市大学新区某大型超市和饲料商店。
分别用超纯水配制质量浓度均为1.0 mg/mL的杆菌肽、多粘菌素B和粘杆菌素标准储备液,于-20 ℃避光保存,使用时用流动相稀释至所需浓度,现用现配。用乙腈溶解并配制10 mmol/L的NBD-F储备液,于-20 ℃避光保存,使用时用乙腈稀释至所需浓度。
1.2 样品前处理
称取5 g牛奶或饲料样品于50 mL离心管中,分别加入20 mL含4% (v/v)乙酸铅和4% (v/v)三氯乙酸的水溶液,超声提取30 min后,以4 000 r/min离心15 min,移取上清液,待净化。
将上述所得样品提取液转移至Oasis HLB固相萃取柱,固相萃取柱分别用6 mL甲醇和水依次活化后上样,首先弃去流出液,然后用6 mL水洗涤,弃去流出液,用6 mL甲醇洗脱,收集洗脱液并于40 ℃氮气吹干。残余样品用1.0 mL流动相溶解混匀,用0.22 μm有机滤膜过滤,然后用于荧光衍生与CEC-LIF分析。
1.3 多肽类抗生素的荧光衍生反应
取3种5 μg/mL的多肽抗生素混合溶液200 μL,加入乙醇200 μL和硼酸缓冲液(pH 7.5, 50 mmol/L)200 μL,再加入200 μL含NBD-F(1 mmol/L)的乙腈溶液,振荡混匀,于60 ℃金属浴中避光衍生反应45 min,冷却至室温,即可得到荧光衍生化样品;另取200 μL乙醇和400 μL硼酸缓冲液(pH 7.5, 50 mmol/L),加入200 μL含NBD-F(1 mmol/L)的乙腈溶液,在同样条件下进行衍生,作为试剂空白。衍生化样品用流动相稀释至所需浓度,用于CEC-LIF分析。
1.4 色谱条件
色谱柱:苯基毛细管电色谱填充柱(100 μm i. d.,总长45 cm,有效长度25 cm, 粒径3 μm,苏州环球色谱有限公司);流动相:乙腈-K2HPO4-KH2PO4缓冲液(10 mmol/L, pH 5.0)(55∶45, v/v);等度洗脱;流速:0.02 mL/min;分离电压:-10 kV;辅助压力:3.8 MPa; LIF检测器波长:λex/λem=473 nm/530 nm。流动相在使用前超声脱气30 min;每天进样前,先用流动相平衡电色谱柱系统,直至获得稳定的电流和基线信号;每次更换流动相也需用流动相平衡至基线稳定。
2 结果和讨论
2.1 荧光衍生反应条件的优化
多数环状多肽类抗生素分子本身没有发色团,紫外吸收较弱,存在D-氨基酸及其对映体,整体结构具有空间立体位阻,结构复杂,且在实际动物性样品中的残留在痕量级,一般的紫外检测器难以实现限量测定。但是PTs结构中含有氨基、羧基和羟基,可以采用荧光衍生。作为新一代的苯并呋咱类荧光试剂,NBD-F相较于NBD-Cl而言,荧光衍生效率高,衍生速度快,可与伯胺、仲胺反应生成具有较强荧光的产物。因此实验选择NBD-F作为PTs的荧光衍生试剂,具体的衍生原理见图1。

图 1 NBD-F衍生含氨基化合物的原理图Fig. 1 Schematic diagram of derivatization for amino-containing compounds with NBD-FNBD-F: 4-fluoro-7-nitro-2,1,3-benzoxadiazole.
2.1.1衍生试剂NBD-F浓度的选择
在进行荧光衍生反应时,需加入适量的衍生试剂来保证目标物反应完全,使衍生效率达到最大。但过量的衍生试剂将增大背景干扰,导致分离效率降低,无法准确定量。研究表明,当NBD-F浓度为0.1~3.0 mmol/L时,随着NBD-F浓度的增加,PTs的衍生效率逐渐提高,并在1.0 mmol/L时达到最大值,表明衍生反应基本完全;继续增大NBD-F浓度,将导致衍生试剂和水解产物残留,荧光效率不再增加。因此选择1 mmol/L的NBD-F进行下一步实验。
2.1.2衍生反应pH值的选择
NBD-F衍生化氨基的反应通常在碱性条件下进行,而多肽类抗生素在弱酸性条件下才能稳定。在50 mmol/L的硼酸缓冲液中,考察了反应介质的pH值在6.5~8.9范围内对多肽类抗生素NBD-F衍生反应的影响。在pH 6.5~7.5范围内,随着pH值的增大,反应体系的碱性增强,增加了NBD-F与PTs亲核反应的概率,荧光响应有显著提高,当pH值为7.5时达到最大;继续增大pH值,此时目标物不稳定易分解,造成衍生效率下降,副产物增多。因此将反应介质的pH值设为7.5。
2.1.3衍生反应时间的选择
适当增加NBD-F衍生的反应时间有助于荧光衍生效率的提高。考察了反应时间对PTs衍生反应的影响。随着衍生反应时间的延长,衍生产物的色谱峰面积不断增加,当反应进行到45 min时,抗生素衍生产物的荧光响应达到最大值,继续延长衍生反应时间容易使衍生试剂水解产物增多,干扰分析。因此将衍生反应时间设为45 min。
2.1.4衍生反应温度的选择
多肽类抗生素在高温下会迅速降解。为了保证分析物的稳定性,试验在45~75 ℃之间考察衍生反应温度对NBD-F衍生效率的影响。当温度较低时,NBD-F与PTs的反应概率较小,荧光衍生效率较低,随着反应温度逐渐加大,衍生效率逐渐增加,60 ℃时,产物的荧光效率达到最大;再增大反应温度会增加荧光分子与溶剂分子相互碰撞淬灭的概率,使荧光量子产率变小,而且温度过高会产生更多的副产物,干扰对目标物的分离分析。因此最终将衍生反应温度设为60 ℃。
2.2 色谱条件的优化
2.2.1色谱柱的选择
多肽类抗生素衍生自氨基酸,结构中的伯胺基团易与传统C18填料中的残余硅醇基发生强烈的相互作用,导致峰拖尾或峰形异常,因此实验采用不含硅醇基的苯基填充色谱柱为固定相,以获得较好的峰形。
实验还发现,CEC分离多肽类抗生素(BAT、PLM-B和CLS)时每种目标物都出现两个色谱峰,分离难度较大。这是由于PTs结构特性使标准品均含有两种主成分,BAT主要含有BAT-A和BAT-B; CLS主要含有CLS-A和CLS-B; PLM-B含有PLM-B1和PLM-B2。Kang等[24]采用CE-UV技术分离BAT、PLM-B和CLS时也报道了这个问题;Kauann等[25]利用LC-MS/MS检测BAT、PLM-B和CLS时也证实了这一点。
2.2.2流动相缓冲盐的选择
分别考察了磷酸盐缓冲液(磷酸钾缓冲液、磷酸钠缓冲液)、三氟乙酸(TFA)缓冲液、2-吗啉乙磺酸(MES)缓冲液对多肽类抗生素荧光衍生产物分离效果的影响。相较于其他缓冲液,磷酸盐尤其是磷酸钾缓冲液对多肽类抗生素的分离具有更好的效果,分析物与残留的衍生试剂NBD-F及水解产物NBD-OH间的分离程度更好。因此选择作为流动相缓冲液。

图 2 缓冲液浓度对多肽抗生素电色谱分离的影响Fig. 2 Effect of buffer concentration on the CEC separation of peptide antibiotics Conditions: mobile phase, acetonitrile (ACN)-phosphate buffer (PB) (pH 5.0) (55∶45, v/v); supplementary pressure, 3.8 MPa; separation voltage, -10 kV; pump flow rate, 0.020 mL/min; detection wavelength, λex/λem=473 nm/530 nm. Peaks: 1 and 2. polymyxin B (PLM-B); 3 and 4. bacitracin (BAT); 5 and 6. colistin (CLS). NBD-OH: 4-fluoro-7-hydroxy-2,1,3-benzoxadiazole.
在毛细管电色谱分离过程中,流动相的主要驱动力来自电渗流、电泳力或压力流,而流动相的黏度影响着电泳迁移率和电渗流的大小。实验采用乙腈-磷酸盐缓冲液(pH 5.0)(55∶45, v/v)为流动相,考察缓冲液浓度(5~20 mmol/L)对3种PTs分离效果的影响(见图2)。结果表明,随着缓冲盐浓度的增大,流动相黏度逐渐变大,扩散系数逐渐变小,使得双电层厚度减小,管壁的Zeta电势也逐渐减小,导致电渗流变小,目标物分析时间延长。其中,BAT(pKa≈5.5)和CLS荧光衍生产物色谱峰的保留时间增加趋势较明显,这可能是由于在pH 5.0时这两种物质带负电荷,电泳方向与电渗流方向相反;多粘菌素B的荧光衍生产物此时更趋向于中性分子,所以色谱峰保留时间主要受电渗流影响,变化略小。综合考虑分析物的保留时间、分离度和峰形,实验选择10 mmol/L磷酸钾盐溶液作为流动相缓冲液。
2.2.3缓冲液pH值的选择
多肽类抗生素的结构中含有两性的氨基酸基团,因此流动相的pH值不仅影响电渗流的大小,也决定了其衍生产物的存在形式,从而影响CEC的分离效率。由于多肽抗生素与NBD-F试剂发生衍生反应后,原先结构中的伯胺、仲胺基团被中性的酰胺键取代,衍生产物的带电情况将主要决定于多肽结构中的羧基基团。考察了流动相缓冲液pH值对目标物分离效果的影响,结果表明,在pH 3.5~6.0范围内,增大pH值有利于抗生素衍生产物结构中羧基的解离,所带负电荷增多,电泳淌度也增加,而苯基毛细管填充柱主要带正电荷,提供阳极电渗流,这使得带负电荷的衍生物在电色谱柱上的流出时间逐渐延长,有利于分离;随着pH值的持续增高(pH >5),衍生产物变得不稳定,虽然分离度略有提高,但是对色谱峰响应与柱效的影响较大。综合考虑分离度、柱效与分析时间,将缓冲液的pH值设为5.0。
2.2.4流动相中有机改性剂的选择
在CEC中,有机改性剂比例的大小影响着流动相的极性,同时也影响分析物的色谱保留行为。通过改变流动相中乙腈的比例,考察其对抗生素衍生产物分离效果的影响(见图3)。结果显示,随着乙腈体积分数的增加,分析物在色谱柱上的保留时间减少,这是由于有机相比例的增加使得流动相的黏度减小,Zeta电位和电渗流变大,降低了目标物在苯基反相柱上的保留。当有机相的体积分数超过55%时,虽然分析物的响应值有明显的增大,但是分离度却有所降低。综合考虑,将流动相中乙腈的体积分数设为55%。
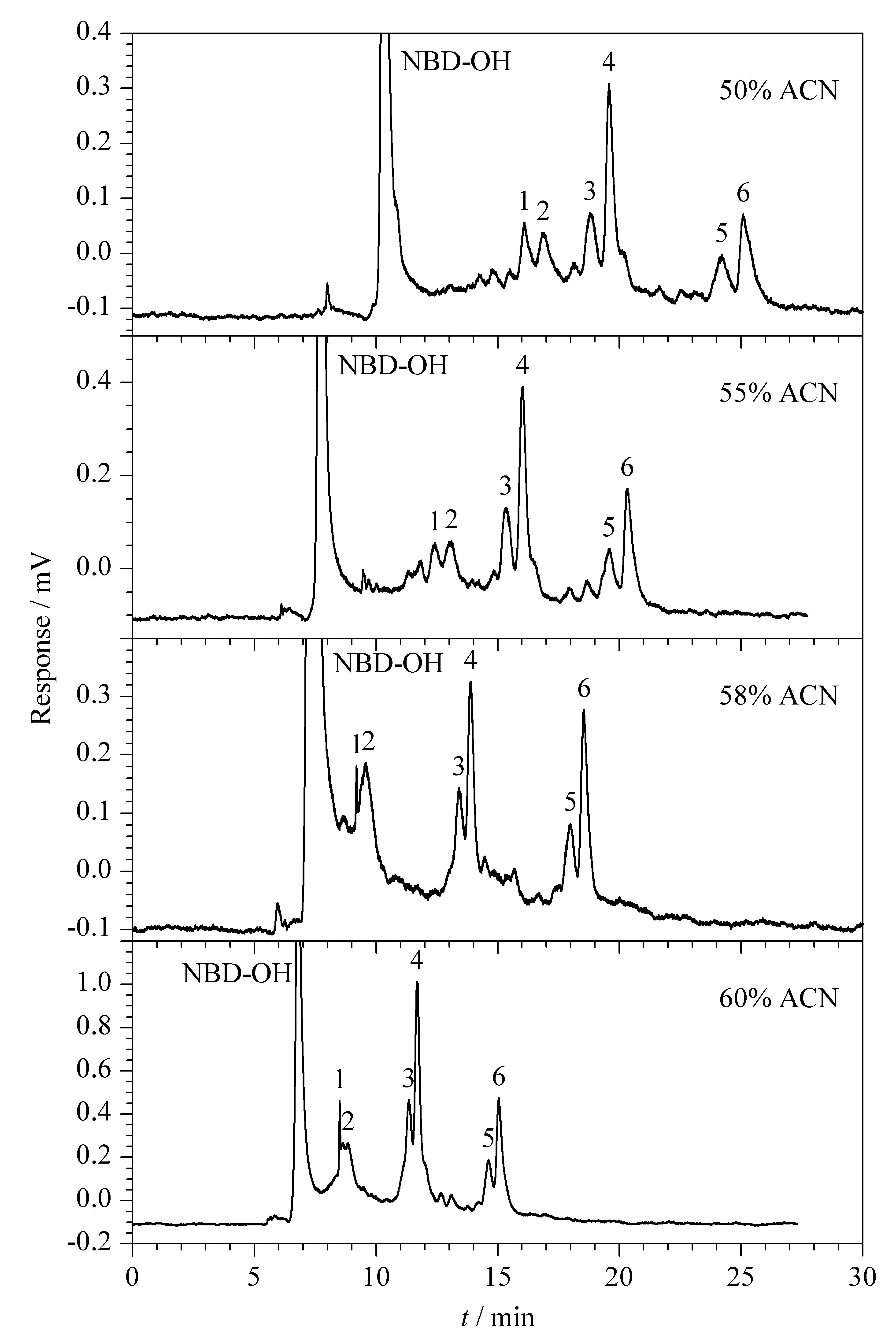
图 3 乙腈的体积分数对3种多肽抗生素分离的影响Fig. 3 Effect of volume fraction of ACN on the separation of the three peptide antibiotics Conditions: mobile phase, ACN-PB (10 mmol/L, pH 5.0); supplementary pressure, 3.8 MPa; applied voltage, -10 kV; pump flow rate, 0.020 mL/min; detection wavelength, λex/λem=473 nm/530 nm. Peaks 1-6 were the same as that in Fig. 2.
2.2.5辅助压力的选择
在CEC分离中引入辅助压力一方面可以有效地抑制气泡产生,防止分离柱在高压电场下断流,提高稳定性;另一方面也能够缩短分析时间。实验通过调节微流控单元中的反压阀,改变施加在毛细管柱端的辅助压力(1.7、3.8、5.2和7.0 MPa),考察其对分析物保留时间和分离度的影响。实验表明,辅助压力增大将缩短样品的分析时间,但分析物的分离度却有所降低,尤其是多粘菌素B1和多粘菌素B2之间的分离度明显降低,且容易受到衍生试剂及其水解峰影响,但是较低的辅助压力会使流动相迁移速度变慢,分析时间延长。因此最终将辅助压力设为3.8 MPa。

图 4 分离电压对多肽类抗生素电色谱分离的影响Fig. 4 Effect of separation voltage on the CEC separation of the peptide antibiotics Conditions: mobile phase, ACN-PB (10 mmol/L, pH 5.0) (55∶45, v/v); supplementary pressure, 3.8 MPa; pump flow rate, 0.020 mL/min; detection wavelength, λex/λem=473 nm/530 nm. Peaks 1-6 were the same as that in Fig. 2.
2.2.6分离电压的影响
实验考察了不同分离电压对PTs衍生产物分离效果的影响。如图4所示,未施加分离电压(0 kV)时,PTs中同一成分不同构型之间(BAT-A和BAT-B、PLM-B1和PLM-B2、CLS-A和CLS-B)由于结构的相似性,无法分离,此时的色谱出峰顺序是BAT-A (BAT-B)、CLS-B (PLM-B1)和CLS-A (PLM-B2)。由于分离驱动力是压力流,分析物按照极性大小的次序流出。随着负向分离电压的施加,在毛细管色谱柱中将产生阳极电渗流,使每一种不同构型的PTs类似物所带电荷产生差异,因此在电场中受到电渗流和电泳力的作用不同,迁移时间发生改变,从而实现结构类似的多肽类抗生素的分离。随着分离电压值的增大,电渗流逐渐变大,分析物的迁移时间明显缩短,每种抗生素类似物的分离度与响应值有所提高,当分离电压为-10 kV时,分析物之间基本能达到基线分离,分离度>1.1。进一步加大分离电压使得基线不稳、噪声变大,分析物之间以及与衍生试剂基质峰的分离度也大大降低。因此实验将分离电压设定为-10 kV。
在上述优化的CEC分离条件下,3种多肽类抗生素的典型电色谱图见图5。

图 5 3种多肽类抗生素的毛细管电色谱图Fig. 5 CEC chromatogram of the three peptide antibioticsPeaks 1-6 were the same as that in Fig. 2.
2.3 线性范围和检出限
配制一系列不同质量浓度的多肽类抗生素混合标准溶液,在最优的色谱条件下进行荧光衍生和CEC-LIF分析。Moralesmunoz和de Castro[26]以邻苯二醛为柱前衍生试剂,采用HPLC-FD方法对饲料中粘杆菌素进行定量分析时指出,对于结构成分复杂的多肽类抗生素,每个抗生素的峰面积可以以标准样品中主要成分的峰面积总和来测定,并用于线性关系的研究。本文以测得的分析物的峰面积和质量浓度绘制工作曲线,得到方法的线性关系和检出限(S/N=3),结果见表1。可以看出,PTs衍生产物的峰面积与各分析物的质量浓度在各自范围内呈良好的线性关系,最低检出限为5.0 ng/mL,可满足多肽类抗生素兽药残留限量的检测要求。该方法的LOD值与文献报道的采用HPLC-MS/MS[27]、HPLC-UV[28]、HPLC-FLD[29]和CE-UV[30]时低1~2个数量级,与结合在线富集技术的CE-MS[31]、HPLC-四极杆/静电场轨道阱高分辨质谱(HPLC-Q/Extractive HRMS)[32]及固相萃取-CZE-ESI-MS的检测水平相当[33]。
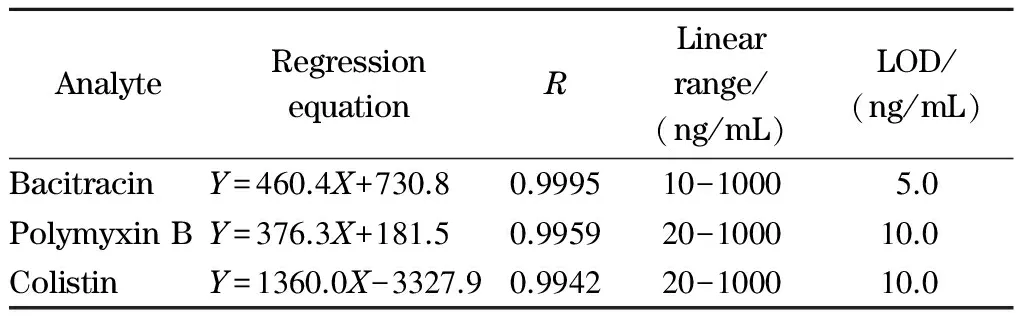
表 1 3种多肽类抗生素的回归方程、相关系数(R)、线性范围和检出限Table 1 Regression equations, correlation coefficients (R), linear ranges and limits of detection (LODs) of the three peptide antibiotics
Y: peak area, μV5s;X: mass concentration, ng/mL.
现有动物性食品中多肽类抗生素如杆菌肽或粘杆菌素的国家标准分析方法[13]均采用LC-MS/MS法,其使用成本与操作要求均较高,检出限为50 ng/mL。本方法无需梯度洗脱,衍生快速高效,检出限更低,而且可以避免大量非荧光杂质的干扰,对于动物性食品中兽用PTs残留的监测具有适用性。
2.4 稳定性和重复性
在最优条件下,对1.0 μg/mL的3种多肽类抗生素混合标准溶液进行荧光衍生和CEC-LIF检测分析,连续测定6 d,每天进样3次,测得3种分析物保留时间的日间和日内相对标准偏差(RSD)分别为8.7%~9.3%和1.2%~1.8%;峰面积的日间和日内RSD分别为4.1%~6.5%和1.7%~3.5%。说明该方法的稳定性和重复性均良好,可用于残留分析研究。
2.5 实际样品分析
将所建立的CEC-LIF分析方法分别用于加标牛奶和饲料样品中多肽类抗生素(0.1 μg/mL)的含量测定(见图6),同时采用国家标准方法[14]检测粘杆菌素,并与本方法进行对比。两种方法均只在空白饲料样品中检测到粘杆菌素,其中采用CEC-LIF时的平均含量为34.68 μg/kg;采用HPLC-MS时的含量为34.96 μg/kg,均未检测出杆菌肽和多粘菌素B。
分别采用CEC-LIF与HPLC-MS/MS在3个添加水平(60、100和200 ng/mL)下对空白牛奶和饲料样品进行加标回收试验,结果见表2。CEC法测得的平均回收率为72.9%~112.4%; HPLC-MS法测得的平均回收率为76.6%~120.9%,相对标准偏差≤9.7%。本方法的精密度和准确度均符合兽药残留限量的分析要求。
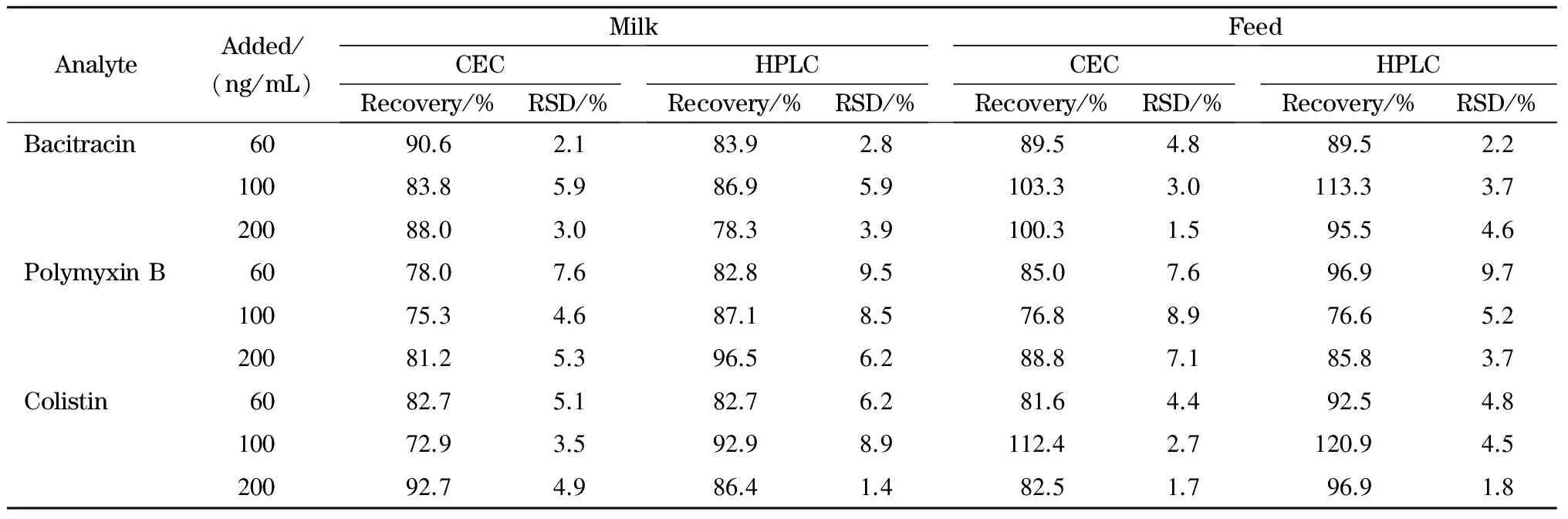
表 2 牛奶和饲料样品中3种多肽类抗生素的加标回收率(n=3)Table 2 Spiked recoveries of the three peptide antibiotics in milk and feed samples (n=3)
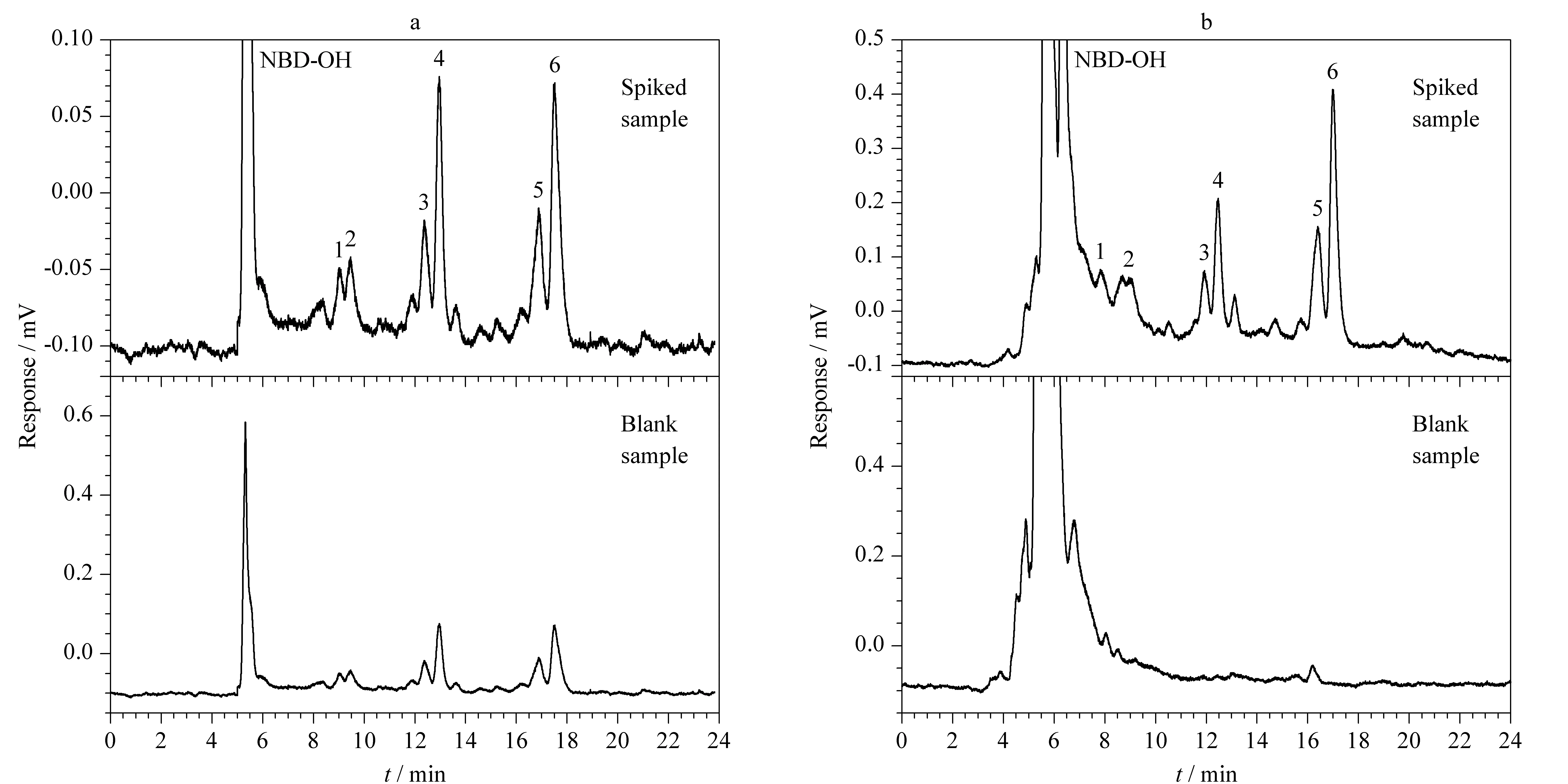
图 6 加标(a)饲料和(b)牛奶样品中3种多肽类抗生素(0.1 μg/mL)的色谱图Fig. 6 Chromatograms of the three peptide antibiotics (0.1 μg/mL) spiked in (a) feed and (b) milk samplesPeaks 1-6 were the same as that in Fig. 2.
3 结论
本文利用毛细管电色谱与激光诱导荧光检测联用技术,发展了环状多肽类抗生素兽药残留的分析方法。建立了基于NBD-F高效衍生化的多肽类抗生素荧光衍生方法,解决了环状多肽结构复杂多样、组分相近、分离选择性差的分析难点。通过调节流动相组成、分离电压与辅助压力,有效地调控了抗生素的分离行为。该法选择性好,分析快速,低耗且预处理简单,为动物源食品中兽用PTs残留分析与评估提供了新的手段。
参考文献:
[1]de la Fuente-Nunez C, Silva O N, Lu T K, et al. Pharmacol Ther, 2017, 178: 132
[2]Roberts K D, Azad M A K, Wang J P, et al. ACS Infect Dis, 2015, 1(11): 568
[3]Liu J J, Jin F, She Y X, et al. Chinese Journal of Analytical Chemistry, 2011, 39(5): 652
刘佳佳, 金芬, 佘永新, 等. 分析化学, 2011, 39(5): 652
[4]Oh Y J, Plochberger B, Rechberger M, et al. J Mol Recognit, 2017, 30(6): e2605
[5]Zakharchenko N S, Pigoleva S V, Yukhmanova A A, et al. Genetika, 2009, 45(8): 929
[6]Park J E, Kim H R, Park S Y, et al. J Appl Microbiol, 2017, 123(5): 1133
[7]Song R, Shi Q Q, Yang P Y, et al. J Funct Foods, 2017, 36: 387
[8]Xin H Y, Ji S Y, Peng J Y, et al. Int J Antimicrob Ag, 2017, 49(4): 427
[9]Boison J O, Lee S, Matus J. Anal Bioanal Chem, 2015, 407(14): 4065
[10]Gorissen B, Reyns T, Devreese M, et al. Anal Bioanal Chem, 2015, 407(15): 4447
[11]Li J S, Qiu Y M, Wang C. Veterinary Drug Residues Analysis. Shanghai: Shanghai Scientific and Technical Publishers, 2002
李俊锁, 邱月明, 王超. 兽药残留分析. 上海: 上海科学技术出版社, 2002
[12]Zhang D, Park J, Kim D, et al. J Sep Sci, 2015, 38(14): 2371
[13]GB/T 22981-2008
[14]Ministry of Agriculture. No. 2086 Bulletin of the Ministry of Agriculture of the People’s Republic of China. (2014-04-01) [2017-10-12]. http://down.foodmate.net/standard/sort/3/40781.html
农业部. 中华人民共和国农业部公告2086号. (2014-04-01) [2017-10-12]. http://down.foodmate.net/standard/sort/3/40781.html
[15]Tsuda T. Anal Chem, 1988, 60(17): 1677
[16]Ma M S, Han H W, Liu G Q, et al. Chinese Journal of Chromatography, 1995, 13(4): 257
马明生, 韩慧婉, 刘国诠, 等. 色谱, 1995, 13(4): 257
[17]Jung M E, Dong T A, Cai X. Tetrahedron Lett, 2011, 42(33): 2533
[18]Omar M A, Hammad M A, Salman B I, et al. Spectrochim Acta A, 2016, 157: 55
[19]Aoyama C, Santa T, Tsunoda M, et al. Biomed Chromatogr, 2004, 18(9): 630
[20]Zhang L Y, Tang X C, Sun M X. J Chromatogr B, 2005, 820(2): 211
[21]Carlucci G, Selvaggi F, Sulpizio S, et al. Biomed Chromatogr, 2015, 29(6): 911
[22]Wu H H, Wang Q Q, Meng R, et al. J Pharmaceu Biomed, 2015, 121: 244
[23]Wu Y M, Lin J, Wu Q Z, et al. J Pharmaceu Biomed, 2010, 53(5): 1324
[24]Kang J, Vankeirsbilck T, Schepdael A V, et al. Electrophoresis, 2000, 21(15): 3199
[25]Kaufmann A, Widmer M. Anal Chim Acta, 2013, 797(40): 81
[26]Moralesmunoz S, de Castro M D. J Chromatogr A, 2005, 1066(1/2): 1
[27]Chiesa L M, Nobile M, Panseri S, et al. Food Chem, 2017, 235: 111
[28]Pfeifer C, Fassauer G, Gerecke H, et al. J Chromatogr B, 2015, 990: 7
[29]Chepyala D, Tsai I L, Sun H Y, et al. J Chromatogr B, 2015, 980: 48
[30]Sautrey G, Duval R E, Chevalley A, et al. Electrophoresis, 2015, 36(20): 2630
[31]Hasan M N, Park S H, Oh E, et al. J Sep Sci, 2010, 33(23/24): 3701
[32]Zhu W X, Yang J Z, Li S, et al. Chinese Journal of Chromatography, 2017, 35(2): 156
祝伟霞, 杨冀州, 李睢, 等. 色谱, 2017, 35(2): 156
[33]Wuethrich A, Haddad P R, Quirino J P. Electrophoresis, 2016, 37(9): 1139
猜你喜欢
河北北方学院学报(自然科学版)(2022年11期)2022-02-03
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
中国科技纵横(2021年24期)2021-03-02
中成药(2018年6期)2018-07-11
现代园艺(2017年13期)2018-01-19
现代检验医学杂志(2016年3期)2016-11-15
国外医药(抗生素分册)(2016年1期)2016-07-10
药学与临床研究(2015年4期)2015-06-05
中国当代医药(2015年22期)2015-03-01
