
黑斑息肉综合征一例基因分析及临床诊治
2018-04-02 08:32:08杨治宇吴庆华信艳萍史惠蓉
郑州大学学报(医学版) 2018年2期
杨治宇 ,吴庆华 ,信艳萍 ,张 毅 ,史惠蓉
1)郑州大学第一附属医院妇科 郑州 450052 2)郑州大学第一附属医院遗传与产前诊断中心 郑州 450052 3)郑州大学附属郑州中心医院 郑州 450052 4)郑州大学第一附属医院生物治疗中心 郑州 450052
黑斑息肉综合征(Peutz-Jeghers syndrome, PJS)又称色素沉着息肉综合征,是一种较罕见的常染色体显性遗传病,主要特征是皮肤黏膜色素沉着及胃肠道多发错构瘤性息肉。PJS可导致肠套叠和肠梗阻等严重的并发症。此外,PJS患者发生恶性肿瘤的风险显著增加,可累及胃肠道、卵巢、乳腺、睾丸和肺脏等[1]。位于19p13.3处的丝氨酸-苏氨酸激酶11(serine/threonine kinase 11,STK11)基因突变是PJS发生的主要原因[2-3]。STK11基因编码蛋白是一种肿瘤抑制因子,是参与细胞增殖、细胞极性、能量代谢和肿瘤发生的多效性细胞因子[4-6]。本研究对1例黑斑息肉综合征的患者进行STK11基因突变检测,回顾性分析其55 a的病史资料及6 a随访资料,以期对该病的临床诊治进行进一步的思索和探讨。
1 对象与方法
1.1研究对象患者,女,63岁,2011年于郑州大学第一附属医院行遗传咨询,自述在外院已临床诊断为PJS,要求了解家族再发风险和生育指导。患者目前口唇及口腔黏膜有少量浅淡的斑点,无贫血貌,余外观发育无异常。经仔细询问,患者母亲和外婆均有口唇黏膜黑斑出现,未曾行胃肠镜检查,外婆曾患子宫内膜癌,其母曾患子宫内膜癌和大网膜腺癌;其27岁的儿子有口唇黏膜黑斑,无明显胃肠道症状;余家族亲属均体健。
1.2STK11基因突变检测经知情同意后,抽取患者外周肘静脉血2 mL,使用全血基因组DNA提取试剂盒提取外周血基因组DNA,行PCR扩增。包括STK11基因所有外显子的引物序列参考文献[7],由上海生工生物工程有限公司合成。反应体系20 μL,包括DNA模板40 ng, 10 μmol/L引物0.5 μL,10 μL Taq PCR Master Mix2×。反应条件:94 ℃预变性5 min,94 ℃变性30 s,62 ℃退火30 s,72 ℃延伸45 s,共40个循环。产物经10 g/L琼脂糖凝胶电泳,紫外线下观察结果。产物经纯化,直接进行双向测序(ABI3130XL测序仪),测序结果应用Chroma 2.0软件分析,采用NCBI BLAST在线软件与GenBank中的正常序列进行比对。
1.3随访通过电话和门诊咨询,每半年对患者的病情询问一次,随访至今。定期行消化道内镜检查并根据情况行内镜息肉手术。
2 结果
2.1STK11基因突变检测结果该患者携带STK11基因c.910C→T杂合突变。突变位点位于第6外显子,cDNA的第910位胞嘧啶突变为胸腺嘧啶,引起第304位氨基酸由苏氨酸替代了丝氨酸(p.R304W)。测序图见图1。
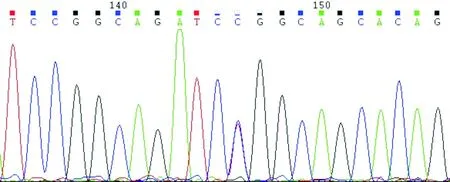
图1 测序图
2.2随访情况患者从3岁起首先出现口唇和口腔颊黏膜黑斑;随年龄增长面积增多且颜色加深,面部、手掌及足掌逐渐出现大小不等、深浅不一的黑色素斑;自23岁起,黑斑症状逐渐减轻,间断排便困难伴大便潜血病史五十余年。患者41 a前因腹痛行剖腹探查,术中见空肠套叠伴扭转,套叠肠管已坏死,行小肠部分切除术,小肠及结直肠内可见散在大量息肉,直径0.3~2.5 cm,病理提示为错构瘤样息肉。18 a前因排便困难行直肠镜检查,切除结肠息肉2枚。自2011年来我院就诊,2011年因腹痛伴黑便行肠镜检查切除结肠息肉80枚;2013年行肠镜检查并切除结肠息肉20余枚,行胃肠镜检查并切除胃体息肉16枚;2014年行胃肠镜检查并切除结肠息肉20余枚;2016年行胃肠镜检查并切除胃体息肉12枚,小肠息肉10枚;目前息肉仍未完全切除。除胃肠道手术外,该患者还曾于33 a前因子宫内膜异位症行子宫次全切术,4 a前因乳房纤维瘤行双乳肿块旋切术。
3 讨论
PJS是常染色体显性遗传病,Peutz于1921年首次对该病进行了介绍,Jeghed在1949年对该病进行了详细的描述。该病主要特征为皮肤黏膜色素沉着,见于95%的PJS患者且是最早出现的症状,好发于口腔及口唇黏膜,其次为手掌、足底,部分患者可见颜面部色素沉着,色斑可在青春期和成年期减退;胃肠道多发错构瘤性息肉,可导致慢性出血及贫血、肠梗阻和肠套叠,需要反复手术和肠切除且易复发;约50%的患者有明确的家族遗传史。依照PJS的诊断标准[8],可根据临床表现、消化道息肉病理学检查和家族史进行诊断。
连锁分析显示,PJS主要致病基因为STK11,又称LKB1基因,长度为23 kb,含有9个外显子,编码一种含433个氨基酸的丝氨酸-苏氨酸激酶。STK11蛋白主要包括3个结构域:N末端非催化域(1~49位氨基酸),催化酶域(49~309位氨基酸)和C末端非催化的调节域(309~433位氨基酸)[2]。94%以上的PJS患者可检测到STK11基因突变,包括点突变和缺失、插入、重复等[3];携带STK11基因突变的个体在遗传性PJS家族中的外显率为100%。本例患者检测到STK11基因c.910C→T(p.R304W)杂合突变,该突变存在于催化酶域Ⅺ(277~309位氨基酸)中;蛋白模型提示R304W突变可破坏自身磷酸化活性,体外实验证明R304W氨基酸改变可导致STK11激酶活性彻底消失[5-6]。
黑斑息肉综合征患者的息肉最常见于小肠,其次为空肠、回肠和十二指肠,少见于胃、大肠和肠外,严重并发症多为胃肠道息肉导致的出血、肠套叠、肠梗阻或癌变。息肉的大小、数量影响患者的临床症状及治疗手段。息肉反复生长于胃、小肠和大肠,数目多达100个以上,目前患者未出现严重并发症,可能与其多年来定期监测和内镜下多次行干预手术有关。
与普通人群相比,PJS患者恶性肿瘤发生风险明显提高,60~70岁患者的癌症风险达37%~93%[9]。大部分肿瘤来源于胃肠道,包括结肠癌、胃癌、小肠癌和胰腺癌,肠外来源的肿瘤有卵巢癌、乳腺癌、睾丸癌和肾癌等[10]。本例的外祖母、母亲均出现口唇黏膜黑斑,未接受任何治疗措施,均因子宫内膜癌去世。与Knudson的二次打击学说相符,PJS患者所患的恶性肿瘤中均发现STK11基因的杂合性缺失[11-12]。STK11基因发挥抑癌作用的机制还不甚清楚,其可能参与了AMPK调控的能量代谢、细胞周期、P53介导的细胞凋亡、Wnt信号通路、TGF-β信号通路、ras介导的细胞转化和细胞极性,以及通过抑制mTOR信号通路调控细胞增殖等[13]。有学者[14-16]认为STK11基因截短突变、外显子3和8突变携带者较其他突变类型携带者发生恶性肿瘤、多发息肉或乳腺癌的风险更高。然而,一些大样本的研究[17-18]提示STK11突变类型与肿瘤风险无关。故PJS患者的基因突变类型与肿瘤发生风险的相关性仍不明确。
目前该病的治疗主要针对消化道息肉和恶性肿瘤的预防和对症治疗。对于PJS患者开始行胃肠息肉筛查的时间仍有不同意见,较统一的专家共识[19-20]建议患者8岁起行内镜下全消化道检查,直径>5 mm的息肉建议切除;发现息肉者需每3 a复查一次;如未检出息肉,则18岁时再次行常规检查;50岁以上的患者因恶性肿瘤发生风险增加,建议每1~2 a复查1次;60岁的PJS患者乳腺癌累积发病风险为31%~54%,建议25~30岁起每年行乳腺MRI或超声检查,50岁起改为乳房X线片检查。目前尚无证据支持需对PJS患者行生殖系统恶性肿瘤或其他类型肿瘤的常规筛查。内镜息肉切除术可以有效降低息肉相关并发症和恶性肿瘤发生风险。尽管在动物实验和临床试验中已观察到mTOR信号通路抑制剂雷帕霉素[21-23]和COX-2抑制剂赛来考西[24]能明显降低PJS患者肠道息肉负荷量,但目前仍未对PJS患者常规使用此类药物治疗。
通过对该病例临床诊治资料的回顾分析,密切监测病情发展并及时处理消化道息肉是控制PJS进展的最佳策略,而对有PJS家族史的生育人群在孕期进行绒毛或羊水取材行STK11基因筛查可以避免患儿出生,是对PJS最有效的预防方法。
[1] RESTA N,PIERANNUNZIO D,LENATO GM,et al.Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients:results of an Italian multicenter study[J].Dig Liver Dis,2013,45(7):606
[2] HANKS SK,QUINN AM,HUNTER T.The protein kinase family:conserved features and deduced phylogeny of the catalytic domains[J].Science,1988,241(4861):42
[3] ARETZ S,STIENEN D,UHLHAAS S,et al.High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome[J].Hum Mutat,2005,26(6):513
[4] RESTA N,SIMONE C,MARENI C,et al.STK11 mutations in Peutz-Jeghers syndrome and sporadic colon cancer[J].Cancer Res,1998,58(21):4799
[5] BOUDEAU J,KIELOCH A,ALESSI DR,et al.Functional analysis of LKB1/STK11 mutants and two aberrant isoforms found in Peutz-Jeghers syndrome patients[J].Hum Mutat,2003,21(2):172
[6] NEZU J,OKU A,SHIMANE M.Loss of cytoplasmic retention ability of mutant LKB1 found in Peutz-Jeghers syndrome patients[J].Biochem Biophys Res Commun,1999,261(3):750
[7] WENG MT,WEI SC.Clinical and genetic analysis of Peutz-Jeghers syndrome patients in Taiwan[J].Cancer Res,2010,70(8):354
[8] KOPACOVA M,TACHECI I,REJCHRT S,et al.Peutz-Jeghers syndrome:diagnostic and therapeutic approach[J].World J Gastroenterol,2009,15(43):5397
[9] VAN LIER MG,WAGNER A,MATHUS-VLIEGEN EM,et al.High cancer risk in Peutz-Jeghers syndrome:a systematic review and surveillance recommendations[J].Am J Gastroenterol,2010,105(6):1258
[10]CHEN HY,JIN XW,LI BR,et al.Cancer risk in patients with Peutz-Jeghers syndrome:a retrospective cohort study of 336 cases[J].Tumour Biol,2017,39(6):1
[11]SU GH,HRUBAN RH,BANSAL RK,et al.Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers[J].Am J Pathol,1999,154(6):1835
[12]GRUBER SB,ENTIUS MM,PETERSEN GM,et al.Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome[J].Cancer Res,1998,58(23):5267
[13]ZHAO RX,XU ZX.Targeting the LKB1 tumor suppressor[J].Curr Drug Targets,2014,15(1):32
[14]SALLOCH H,REINACHER-SCHICK A,SCHULMANN KA,et al.Truncating mutations in Peutz-Jeghers syndrome are associated with more polyps, surgical interventions and cancers[J].Int J Colorectal Dis,2010,25(1):97
[15]WANG ZQ,WU BP,MOSIG RA,et al.STK11 domain Ⅺ mutations:candidate genetic drivers leading to the development of dysplastic polyps in Peutz-Jeghers syndrome[J].Hum Mutat,2014,35(7):851
[16]TAN H,MEI LB,HUANG YR,et al.Three novel mutations of STK11 gene in Chinese patients with Peutz-Jeghers syndrome[J].BMC Med Genet,2016,17(1):77
[17]HEARLE N,SCHUMACHER V,MENKO FH,et al.STK11 status and intussusception risk in Peutz-Jeghers syndrome[J].J Med Genet,2006,43(8):e41
[18]VAN LIER MG,WESTERMAN AM,WAGNER A,et al.High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome[J].Gut,2011,60(2):141
[19]BEGGS AD,LATCHFORD AR,VASEN HF,et al.Peutz-Jeghers syndrome: a systematic review and recommendations for management[J].Gut,2010,59(7):975
[20]SYNGAL S,BRAND RE,CHURCH JM,et al.ACG clinical guideline:genetic testing and management of hereditary gastrointestinal cancer syndromes[J].Am J Gastroenterol,2015,110(2):223
[21]KUWADA SK,BURT R.A rationale for mTOR inhibitors as chemoprevention agents in Peutz-Jeghers syndrome[J].Fam Cancer,2011,10(3):469
[22]WEI CJ,AMOS CI,ZHANG NX,et al.Chemopreventive efficacy of rapamycin on Peutz-Jeghers syndrome in a mouse model[J].Cancer Lett,2009,277(2):149
[23]WEI C,AMOS CI,ZHANG N,et al.Suppression of Peutz-Jeghers polyposis by targeting mammalian target of rapamycin signaling[J].Clin Cancer Res,2008,14(4):1167
[24]UDD L,KATAJISTO P,ROSSI DJ,et al.Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2[J].Gastroenterology,2004,127(4):1030
猜你喜欢
当代水产(2022年2期)2022-04-26 14:25:48
中国中西医结合皮肤性病学杂志(2020年4期)2020-09-10 01:38:26
青年文学家(2020年22期)2020-08-31 01:58:41
当代水产(2020年4期)2020-06-16 03:22:52
当代水产(2020年4期)2020-06-16 03:22:46
当代医药论丛(2017年22期)2017-04-12 06:30:37
中国美容医学(2017年1期)2017-03-23 13:28:39
西藏科技(2015年12期)2015-09-26 12:13:45
女士(2015年6期)2015-05-30 20:22:32
意林·少年版(2011年17期)2011-07-06 00:22:36
