
特发性肺间质纤维化与COPD合并肺间质纤维化患者肺功能比较
2018-04-02 06:19徐亚莉陈映红
现代仪器与医疗 2018年1期
徐亚莉 陈映红
特发性肺间质纤维化(Idiopathic pulmonary fibrosis,IPF)是一种慢性炎症性间质性肺疾病,患者以弥漫性肺泡炎、肺泡单位结构紊乱、肺间质纤维化为主要病理改变;目前全球患慢性阻塞性肺疾病(Chronic obstructive pulmonary disease,COPD)人数高达 3.4亿[1-2]。随着COPD加重可并发肺间质纤维化(Pulmonary fibrosis,PF),且COPD向PF的进展过程意味着肺功能出现不可逆转的减退[3-5]。因此,了解IPF与COPD合并PF(PF-COPD)的关系,对于指导临床诊断与治疗、抑制COPD向PF进展均具有重要意义。为此,本文进行了对照分析如下。
1 资料与方法
1.1 一般资料
将2014年3月—2017年3月来院就诊的89例IPF患者及64例PF-COPD患者分别纳入IPF组、PF-COPD组。排除同时合并其他肺部疾病者,入组前对此次研究知情同意。IPF组男47例,女42例,年龄25~71岁,平均(56.13±8.26)岁;PF-COPD组男35例,女29例,年龄27~75岁,平均(55.74±9.02)岁。两组患者年龄、性别比例比较,差异无统计学意义(P>0.05),具有可比性。此次临床研究已征得我院医学伦理委员会批准。
1.2 研究方法
两组均完成X线胸片及胸部CT/HRCT检查;肺功能检测使用Vmax229型肺功能测试系统(深圳市森迪斯科技有限公司),检测指标包括用力肺活量(FVC)、第一秒用力呼气量(FEV1)、第一秒用力呼气量占用力肺活量百分比(FEV1%)及一氧化碳弥散量(DLCO[6]。
1.3 统计学分析
对本临床研究的所有数据采用SPSS18.0进行分析,临床表现、影像学检查结果等计数资料以(n/%)表示,并采用χ2检验,年龄、血气指标、肺功能等计量资料以(±s)表示,并采用t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 临床表现
PF-COPD组桶状胸、肺界下移、湿啰音、肺心病体征发生率高于IPF组,差异有统计学意义(P<0.05)。见表1。
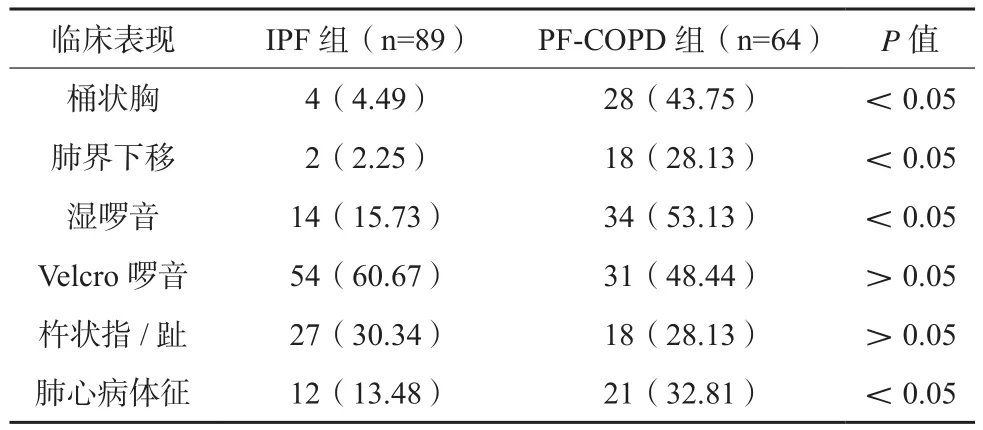
表1 两组患者临床表现比较(n/%)
2.2 影像学表现
X线胸片PF-COPD组胸廓增大、肺透亮度增高、肺纹理紊乱发生率高于IPF组;CT/HRCT检查显示,PF-COPD组肺大泡、肺泡壁破坏发生率高于IPF组,差异有统计学意义(P<0.05)。见表2。

表2 两组影像学检查结果比较(n/%)
2.3 肺功能
PF-COPD组FVC高于IPF组,其FEV1低于后者,PF-COPD组PaCO2高于IPF组差异有统计学意义(P<0.05)。见表3。
表3 两组患者肺功能比较(±s)

表3 两组患者肺功能比较(±s)
指标 IPF组(n=89) PF-COPD组(n=64) P值FVC(%) 63.25±8.04 75.53±8.44 <0.05 FEV1(%) 63.26±9.92 52.26±10.01 <0.05 FEV1% 101.59±11.83 68.36±12.57 >0.05 DLCO(%) 47.26±7.15 53.26±14.41 >0.05 PaO2(mmHg) 67.31±9.95 70.26±9.81 >0.05 PaCO2(mmHg) 36.25±3.04 44.01±7.52 <0.05
3 讨论
COPD和IPF均是临床常见的慢性进行性呼吸系统疾病,近年来,越来越多的研究发现,肺间质纤维化是COPD发展过程中的一种常见病理改变,即随着患者病程进展,病灶逐渐深入肺组织深部,并引发肺间质和肺泡纤维化[7]。PF-COPD的发生不仅可严重破坏肺组织、加重缺氧和弥散功能障碍,还可导致肺动脉高压发生风险上升,进一步加速COPD病程进展[8-9]。因此,了解PF-COPD的临床特征,可指导早期治疗。
既往研究发现,IPF与PF-COPD患者临床表现具有相似性,给临床诊断带来了一定困难[10]。此次研究对比了IPF患者与PF-COPD患者临床表现,结果发现,除两种疾病共有的Velcro啰音、杵状指/趾症状外,PF-COPD患者桶状胸、肺界下移、湿啰音、肺心病体征发生率更高,考虑与PF-COPD患者呼吸困难的进行性加重有关[11-12]。在影像学表现的对比中,可以发现,与IPF患者相比,PF-COPD患者X线胸片胸廓增大、肺透亮度增高、肺纹理紊乱发生率更高,且CT/HRCT图像肺大泡、肺泡壁破坏发生率也明显升高,其原因可能为:除COPD引发的阻塞性通气功能障碍外,PF-COPD导致的限制性通气功能障碍可导致肺部出现大量散在片状炎症影,而急性炎症的存在,也使得肺泡内渗出物增加,进而表现为肺透亮度的升高、肺大泡的增多[13];此外,部分患者可出现肺组织瘢痕形成、皱缩等病理改变,可进一步导致肺泡壁破坏、融合,以及毛细血管闭塞,从而引发肺泡外膜纤维性增厚,形成恶性循环[14]。
在肺功能的对比中,可以发现,较IPF患者而言,PFCOPD患者FVC较高但FEV1偏低,这一结果再次印证了PF-COPD患者同时存在两种疾病的特点,即阻塞性通气功能障碍、限制性通气功能障碍共同存在[15],其原因一方面与COPD所致气道重塑、肺气肿引发的肺组织弹性回缩力下降有关,另一方面,严重的肺间质纤维化所致弥散功能下降也是导致病情持续进展的主要原因[16]。对比两组患者血气指标,可以发现,PF-COPD患者处于更为严重的二氧化碳潴留状态,其原因可能包括[17]:1)肺泡的纤维化改变可加剧肺气肿对终末气道的压迫;2)外周细支气管纤维化对小气道的影响;3)持续的严重低氧血症造成二氧化碳排出受限。由于目前临床尚无针对PF-COPD的标准治疗方案,多数学者均主张实施吸氧、抗感染、止咳化痰、解痉平喘等综合治疗策略,且不建议应用激素和免疫抑制剂。但也有学者发现,糖皮质激素与免疫抑制剂联用可使部分PFCOPD患者病情得到明显改善甚至获得痊愈[18],因此,关于PF-COPD的治疗方案选择仍有待进一步研究。
综上所述,除临床表现、影像学特征外,IPF与PFCOPD患者肺功能及血气指标也存在一定差异,可据此判别IPF与PF-COPD、早期明确COPD患者的肺纤维化改变,从而指导个体化治疗方案,尽可能改善患者预后质量。
[1] RICHELDI L, DU BOIS R M, RAGHU G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis[J]. N Engl J Med, 2014, 370(22): 2071-2082.
[2] KING JR T E, BRADFORD W Z, CASTRO-BERNARDINI S,et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis[J]. N Engl J Med, 2014, 370(22): 2083-2092.
[3] MOLYNEAUX P L, COX M J, WILLIS-OWEN S A G, et al.The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis[J]. Am J Respir Crit Care Med,2014, 190(8): 906-913.
[4] 贺琛, 俞小卫. 慢性阻塞性肺疾病合并肺间质纤维化37例临床分析[J]. 国际呼吸杂志, 2016, 36(5): 350-352.
[5] WIELSCHER M, VIERLINGER K, KEGLER U, et al.Diagnostic performance of plasma DNA methylation profiles in lung cancer, pulmonary fibrosis and COPD[J]. EBioMedicine,2015, 2(8): 929-936.
[6] KUWANO K, ARAYA J, HARA H, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis(IPF)[J]. Respir Investig, 2016, 54(6): 397-406.
[7] IDIOPATHIC PULMONARY FIBROSIS CLINICAL RESEARCH NETWORK. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis[J]. N Engl J Med, 2014, 2014(370):2093-2101.
[8] 潘天宇, 郭忠良, 鲁立文,等. 支气管哮喘-慢性阻塞性肺疾病重叠综合征患者血浆纤维蛋白原变化及其与肺功能的相关性分析[J]. 疑难病杂志, 2017, 16(5):469-472.
[9] SELMAN M, PARDO A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model[J]. Am J Respir Crit Care Med, 2014,189(10): 1161-1172.
[10] MATSUDA T, TANIGUCHI H, ANDO M, et al. COPD Assessment Test for measurement of health status in patients with idiopathic pulmonary fibrosis: A cross-sectional study[J].Respirology, 2017, 22(4): 721-727.
[11] EVANS C M, FINGERLIN T E, SCHWARZ M I, et al.Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways[J]. Physiol Rev, 2016, 96(4): 1567-1591.
[12] HOFFMANN J, WILHELM J, MARSH L M, et al. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension[J].Am J Respir Crit Care Med, 2014, 190(1): 98-111.
[13] FANER R, ROJAS M, MACNEE W, et al. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis.[J]. Am J Respir Crit Care Med, 2012,186(4):306-13.
[14] TOMASSETTI S, GURIOLI C, RYU J H, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis[J].Chest, 2015, 147(1): 157-164.
[15] SOHAL S S, MAHMOOD M Q, WALTERS E H. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer[J]. Clin Transl Med, 2014, 3(1): 33.
[16] 陈永春, 王天轶. 不同时期慢性阻塞性肺疾病患者血清炎性因子水平和肺功能情况分析[J]. 解放军医药杂志, 2017, 29(4):38-40.
[17] WUYTS W A, ANTONIOU K M, BORENSZTAJN K, et al.Combination therapy: the future of management for idiopathic pulmonary fibrosis?[J]. Lancet Respir Med, 2014, 2(11): 933-942.
[18] JOHANNSON K A, VITTINGHOFF E, LEE K, et al. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure[J]. Eur Respir J, 2014, 43(4): 1124-1131.
猜你喜欢
传染病信息(2022年3期)2022-07-15
现代临床医学(2021年4期)2021-07-31
肝博士(2021年1期)2021-03-29
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
老年医学与保健(2017年6期)2017-02-06
医学研究杂志(2015年6期)2015-07-01
中国当代医药(2015年23期)2015-03-01
中国中医药现代远程教育(2014年23期)2014-03-01
中国中医药现代远程教育(2014年13期)2014-03-01
