
肝细胞癌的全基因组关联研究进展*
2018-03-29 01:09吴叶娇张金坤周春晓陈卫昌综述审校
重庆医学 2018年7期
吴叶娇,张金坤,周春晓,陈卫昌 综述,余 强Δ 审校
(1.南京医科大学附属苏州医院/苏州市立医院消化内科 215002;2.苏州大学第一附属医院消化内科,江苏苏州 215006)
肝细胞癌(hepatocellular carcinoma,HCC)起源于构成肝脏实质的肝细胞,是原发性肝癌的最常见组织学类型,约占原发性肝癌的85%~90%。HCC是世界范围内最常见的恶性肿瘤之一,分别居男性恶性肿瘤的第5位和女性恶性肿瘤的第7位[1]。HCC的发病率在全球范围内的分布并不均匀,绝大多数出现(大于80%)在撒哈拉以南的非洲和东亚地区;中国的年龄标准化发病率男性达37.4/10万,女性13.7/10万[2],仅中国的病例数就在全球所有HCC患者中占大约50%[3]。然而在北美洲、南美洲及欧洲,HCC的发病率相对较低。这种显著的差异是由多种因素造成的。HCC涉及多重遗传和环境因素的共同作用[4]。HCC患者预后差,平均3年生存率仅为13%~21%[5]。如此高的全球疾病负担使得识别HCC高危个体和可控的危险因子显得尤为重要。已知数个增加HCC风险的环境因素,如乙型肝炎病毒(hepatitis B virus,HBV)或丙型肝炎病毒(hepatitis C virus,HCV)的感染、接触黄曲霉毒素和大量饮酒、非酒精性脂肪性肝病、糖尿病和血色病[3,6-7]。然而,人群中仅一小部分暴露于这些危险因素的个体最终罹患HCC,这充分突显出遗传易感性是另一个影响HCC发生、发展的重要因素。
目前,血清甲胎蛋白水平测定和肝脏的影像学检查是高危人群筛查的主要手段。然而,这两种技术的灵敏度较低,使得它们的有效性受到限制。此外,它们并非旨在识别高危个体,而是已经罹患HCC的患者。未来的医学实践将紧紧围绕4P概念,即预测性(predictive)、个体化(personalized)、抢占性(preemptive)和实践性(participatory)[8]。其实,在此框架内,目前尚不存在与之相符的HCC遗传标记。因此,与HCC风险增高相关的分子标记将会成为识别高危个体策略中的一种非常有价值的上游工具,自然也会在预防性干预中扮演特殊的角色。
随着基因组学领域的快速发展,整个人类基因组序列完成,二代测序技术出现,基因检测成本相对降低,以及新分析工具应用,让HCC研究领域出现重大的革新性改变。这些进展使得对基因组中成千上万的单核苷酸多态性(single nucleotide polymorphisms,SNPs)进行快速基因型分型成为可能,这就直接促成了全基因组关联研究(genome-wide association study,GWAS)的出现和发展。随之,许多与HCC有关联的遗传易感性位点通过GWAS被发现,为HCC的发病机制研究提供了新的重要线索。本文系统介绍GWAS应用于HCC发病机制研究的最新进展。
1 全基因组关联研究
候选基因策略曾经最为广泛地应用于研究复杂性疾病发生、发展和治疗效果的遗传因素。这种方法是有假设前提的,并因依赖于对候选基因在生理、生物化学或功能等方面的先验知识。因此候选基因策略受到极大的限制。这种局限性导致信息瓶颈而往往引起已报道实验结果缺乏一致性。但GWAS的出现彻底打破了这一局限。
GWAS是一种利用人类全基因组高通量测序和分型的技术,对研究对象的整个基因组中的SNPs进行分型。通常,其研究对象有超过数十万个SNPs,传统技术难以快速测序,但GWAS可以做到。同时,GWAS利用生物统计学和生物信息学的方法,检验SNPs与复杂疾病或特定遗传性状的关联性,由此全面地揭示与疾病发生、发展及治疗相关的遗传位点。GWAS的理论来源于人类常见的复杂性疾病,主要是以常见的等位基因遗传变异为遗传基础[9]。通常,GWAS是利用覆盖人类基因组的高密度SNPs来寻找目标人群(有复杂性疾病或特定遗传性状者)和对照人群之间等位基因频率的差异。如果差异存在统计学意义,就提示在其基因组相应区域可能存在具有潜在功能并且与所研究的复杂性疾病或特定遗传性状有关联的DNA序列变异。随后在新的标本中将继续进行验证这种有统计学意义关联是否仍然存在[10]。
GWAS一般有如下4个步骤[11]:(1)研究设计和准备阶段。包括查阅相关文献、提出科研假说、进行科学研究设计、招募合格的研究对象和对照个体、问卷调查和采集生物学标本。(2)实验室基因分型阶段。包括制备DNA、准备芯片及基因分型和实验室的质量控制等。(3)数据分析阶段。包括调查资料的整理汇总、基因分型资料整理汇总[如缺失值的控制、最小等位基因频率的控制、Hardy-Weinberg连锁不平衡(linkage disequilibrium,LD)的检验、亲属关系的鉴别等][12]、人群分层分析(鉴别种族和调整人群的混杂效应)、各遗传位点与复杂性疾病或特定遗传性状的效应分析,进而分析每个基因、每条通路与复杂性疾病或特定遗传性状的关联,以及基因-环境和基因-基因的交互作用。(4)重复验证阶段。对新发现的易感位点或基因,在同类人群和(或)其他种族人群中进行进一步验证,评价该研究结果的可重复性,以减少假发现率的产生[13]。
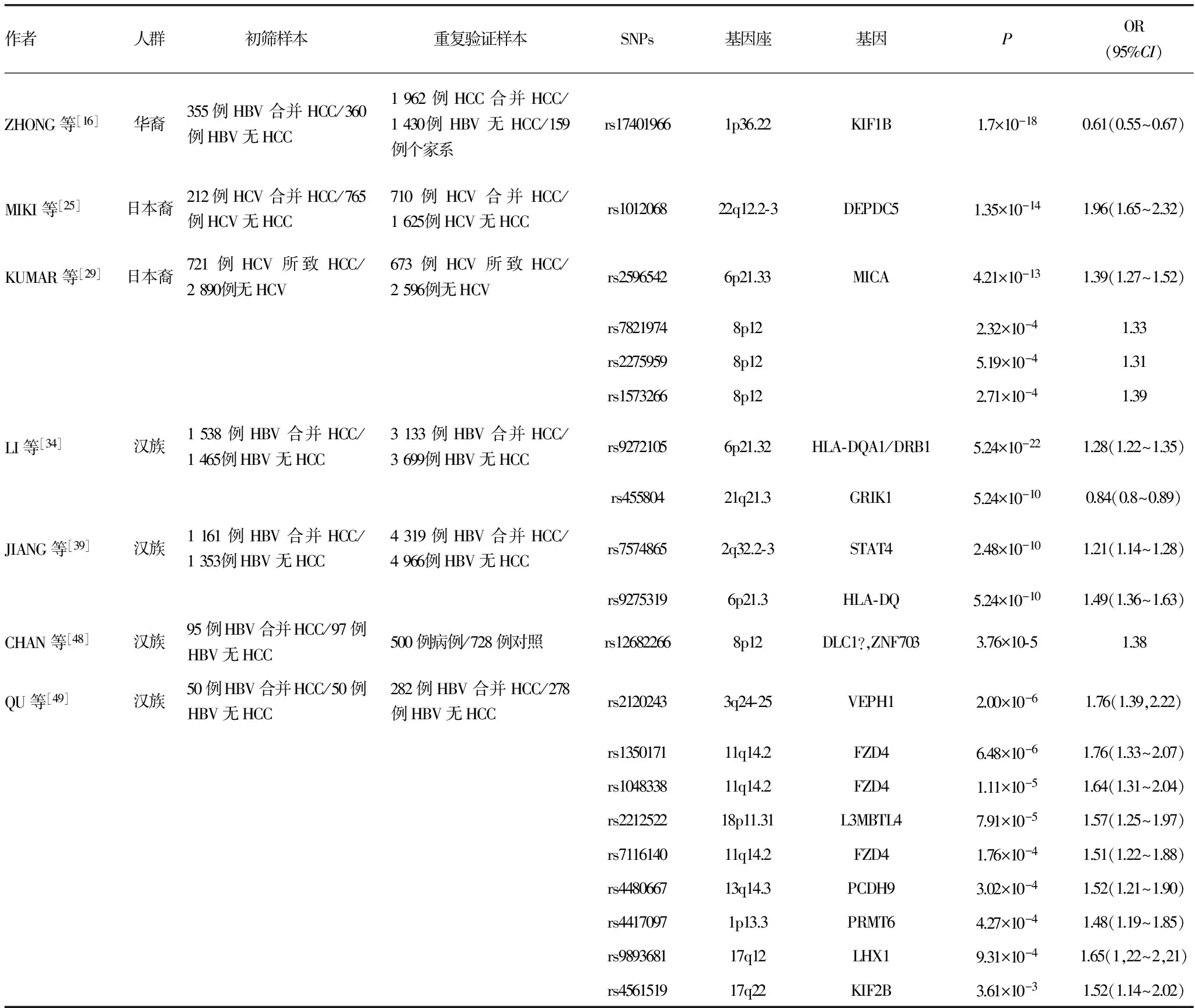
表1 GWAS报道的HCC相关遗传易感位点
1996年GWAS由RISCH等[14]提出,总结了GWAS所需的技术和统计学分析方法,并指出当时GWAS的主要缺乏技术支持而非统计学分析方法。1990年人类基因组计划启动,旨在完成人类基因组测序图。第一阶段的成果公布于2001年。同年,另一个标志性项目启动,即国际人类基因组单体型图计划。该研究记录了人类基因组中大量共同的SNPs并确定了LD的模式和这些变异等位基因之间的关联性。该研究为商业性基因型分型芯片奠定了选择遗传标志物的基础并使日后的GWAS工作进展顺利。2005年Science杂志发表了有关GWAS的文章[15]。自此,与GWAS相关的文献发表量逐年增加,几乎覆盖了所有复杂性疾病和特定遗传性状的表型,报道了许多与复杂性疾病和特定遗传性状有统计学关联的SNPs,并在不同种族的或更大的人群中得到验证。由此可见,GWAS已经成为目前人类复杂性遗传性状和疾病的遗传易感性的主要研究策略。
2 GWAS与HCC
目前,GWAS所发现的与HCC可能相关的遗传易感位点已经达到20个,见表1。其中,针对部分遗传易感位点已经开展了相关生物学功能研究,结果提示部分遗传易感位点在HCC的发生和发展过程中可能发挥重要的作用。本文针对数个遗传易感位点的GWAS结果和致病机制进行综述。
2.1KIF1B 2010年ZHANG等[16]在华裔人群中采用Affymetrix的人类全基因组SNP芯片对355例HCC合并慢性HBV携带的患者和360例慢性HBV携带但无HCC的对照患者进行初筛。在440 794个标记SNPs中发现,在染色体1p36.22上KIF1B基因内的rs17401966变异与乙型肝炎后HCC显著相关。随后在5个独立人群中分别进行了重复验证。这5次验证的研究对象总人数达1 962例HCC合并慢性HBV携带的患者、1 430例慢性HBV携带但无HCC对照患者以及159个家系,均为华裔。经上述6次研究证实该位点与乙肝后HCC的发病有统计学关联(6次研究合并P=1.7×10-18,OR=0.61,95%CI:0.55~0.67)。然而,之后的研究并未得出相同的结论。AL-QAHTANI等[17]报道,在KIF1B基因内的rs17401966变异和乙肝后HCC之间无显著的相关性。SAWAI等[18]在日本人的一项队列研究中也并未发现该两者的相关性。这些大相径庭的结果可能部分因为不同研究人群的遗传构架差异。另一个可能的原因是忽略了在HCC的发生和发展过程中存在复杂的基因-环境交互作用。近期CHEN等[19]在中国人群的一项研究表明KIF1B基因内的rs17401966变异与饮酒之间存在基因-环境交互作用并在HCC的发生、发展过程中发挥重要作用。
1p36的杂合性缺失(loss of heterozygosity,LOH)是多种肿瘤中的常见遗传缺陷,包括HCC[20-22]。1p36.21-36.22内的LOH可能是肝脏癌变的初始事件之一,这意味着该区域可能存在HCC的抑癌基因[22]。rs17401966处在一个224 kb的LD模块内,该模块含UBE4B的3′端,KIF1Bα和PGDβ。KIF1B是一个驱动蛋白超家族成员,编码两个选择性剪接的亚型,KIF1Bα和KIF1Bβ;这两个亚型形成同源二聚体,运输线粒体和突触囊泡前体。KIFs的下调对某些脑部、结肠、乳腺的肿瘤具有致癌作用[23]。两个独立的研究均证实KIF1B可能是成细胞纤维瘤的一个潜在抑癌基因,作用于EglN3脯氨酸羟化酶的下游诱导细胞凋亡[23-24],最终导致恶变和进展得到抑制。所以KIF1B很可能是一个抑癌基因。
2.2DEPDC5 2011年MIKI等[25]在日本人中采用Illumina HumanHap芯片对212例慢性HCV合并HCC感染的患者和765例慢性HCV感染但无HCC的对照患者进行初筛。在467 538个SNPs中,发现在染色体22q12.2-3上DEPDC5基因内的rs1012068与丙型肝炎后HCC显著相关。随后在710例慢性HCV合并HCC感染的患者和1 625例慢性HCV感染但无HCC的对照患者中进行独立的重复验证,经调整性别、年龄和血小板计数后证实,该位点与丙肝后HCC的发病有统计学关联(合并P=1.35×10-14,OR=1.96,95%CI:1.65~2.32)。虽然曾有报道称DEPDC1影响膀胱癌的发生[26],该基因含有与DEPDC5类似的DEP结构域,但是DEPDC5的功能目前仍然未知。
近期BURZA等[27]在欧洲裔人群中进行的病例对照研究并未发现DEPDC5基因内的rs1012068变异与HCC的相关性。但研究者注意到该变异与中至重度纤维化相关。肝硬化,是纤维化最严重的等级,也是HCC的主要危险因素[28]。在MIKI等[25]的研究中,大部分HCC患者很有可能同时存在肝硬化。然而对照组的患者并非丙型肝炎后肝硬化而无HCC的患者。所以,MIKI等[25]找到的关联性可能实际上是DEPDC5基因内的rs1012068与严重肝纤维化相关而非HCC。为进一步明确分子机制,BURZA等[27]对肝星状细胞(LX-2)进行了体外研究,结果表明DEPDC5基因的下调导致catenin表达及其下游目标基质金属蛋白酶2(matrix metallopeptidase 2,MMP2)的产生量有所增加。而MMP2是一个与肝脏纤维化密切相关的酶。
2.3MICA 2011年KUMAR等[29]在日本裔中采用Illumina HumanHap芯片对721例HCV所致HCC的患者和2 890例无慢性HCV感染的对照者进行初筛。随后在673例HCV所致HCC的患者和2 596例无慢性HCV感染的对照者中进行重复验证。在432 703个常染色体SNPs中,染色体6p21.33上的MICA基因内的rs2496542与丙肝后HCC的发病显著相关(合并P=4.21×10-13,OR=1.39,95%CI:1.27~1.52)。
MICA基因编码一个膜结合蛋白,是自然杀伤(natural killer,NK)细胞表面的自然杀伤细胞组2D(natural killer cell group 2D,NKG2D)的配体。这些NK细胞通过分泌穿孔蛋白和颗粒酶及死亡受体信号发挥细胞毒性作用,也释放炎症细胞因子以起到抗病毒和对感染和肿瘤免疫应答的效果[30]。因此MICA/NKG2D系统是免疫监视的有效机制。病毒通过破坏MICA的产生以拮抗MICA-NKG2D系统的抗肿瘤作用。而rs2596542正处于MICA转录起始位点的上游。肿瘤细胞也逐渐形成了通过脱落酶使MICA从细胞表面脱落,拮抗MICA/NKG2D激活的信号[31],从而尽可能减少或躲避NKG2D介导的应答机制。血清sMICA的水平在HCC晚期的患者中升高,且与NKG2D表达的下调和NK细胞活性下降相关[32]。这些实验结果均表明NK细胞的功能障碍在逃脱肿瘤监视系统中扮演重要角色[33]。
2.4HLA-DQA1/DRB1与GRIK1 2012年LI等[34]在华裔中对1 538例HCC合并慢性HBV携带的患者和1 465例慢性HBV携带但无HCC的对照患者进行初筛。随后在总人数为3 133例HCC合并慢性HBV携带的患者和3 699例慢性HBV携带但无HCC的对照患者中进行两次独立的重复验证。结果在523 663个常染色体SNPs中,发现染色体6p21.32上的HLA-DQA1/DRB1基因(功能性研究在其后的HLA-DQ基因中共同进行阐述)内的rs9272105变异和染色体21q21.3上的GRIK1基因内的rs455804变异与乙肝后HCC的发病显著相关(合并P=5.24×10-22,OR=1.28,95%CI:1.22~1.35;P=5.24×10-10,OR=0.84,95%CI:0.80~0.89)。
GRIK1编码GLUR5,是一种促离子型谷氨酸受体,作为配体激活通道的亚基参与谷氨酸信号。谷氨酸通过多种分子机制在神经胶质瘤的恶性表型中起关键作用[35]。抑制谷氨酸的释放和(或)谷氨酸受体活性可以有效减少乳腺癌、喉癌和胰腺癌的肿瘤细胞增殖和(或)侵袭[36-38]。LI等[34]研究更进一步验证了谷氨酸信号通路在癌症发生和发展中的重要作用,同时对乙型肝炎后HCC的机制研究具有指导意义。
2.5STAT4与HLA-DQ 2013年JIANG等[39]采用Illumina芯片在中国人群中对1 161例HCC合并慢性HBV携带的患者和1 353例慢性HBV携带但无HCC的对照患者进行初筛。随后在总人数为4 319例HCC合并慢性HBV携带的患者和4 966例慢性HBV携带但无HCC的对照患者中进行两个阶段的重复验证。研究结果显示,在568 280个常染色体SNPs中,染色体2上STATA基因内的rs7574865变异和染色体6上的HLA-DQ基因内的rs9275319变异与乙型肝炎后HCC的发病显著相关(合并P=2.48×10-10,OR=1.21,95%CI:1.14~1.28;P=2.72×10-17,OR=1.49,95%CI:1.36~1.63)。
信号转导及转录激活子(signal transducers and activators of transcription,STAT)可调节造血过程并通过抑制或诱导生长因子和特定的细胞因子影响肿瘤细胞与其免疫微环境的相互作用[40-41]。其中,STAT4基因在免疫反应的过程中扮演重要的角色。由抗原呈递细胞(antigen-presenting cells,APCs)或具有类似APCs功能的细胞分泌的白细胞介素-12激活STAT4;活化的STAT4能促进像NK细胞等的炎症细胞分泌干扰素(interferon,IFN)-γ[42]。IFN-γ是干细胞凋亡、肝脏再生、病毒控制和抑制肿瘤的关键细胞因子。STAT4表达的不平衡能够通过炎性反应、T细胞分化的调控和IFN-γ导致自身免疫性疾病或肿瘤[43]。这些都表明STAT4可能参与HCC发生和发展的病理过程。
HLA系统是编码人类主要组织相容性复合体的基因座的名称。这个超级基因座包含大量与人类免疫系统功能相关的基因。HLA Ⅱ类分子包括3个同种型:HLA-DR、HLA-DQ和HLA-DP。已有大量文献报道了它们与HCC之间的相关性,而LI等[34]和JIANG等[39]有关GWASs的研究从另一个角度再次证实了这一点。rs9272105位于HLA-DQA1和HLA-DRB1之间,而rs9275319位于HLA-DAB1和HLA-DQA2之间。HLA-DQ和-DR编码的蛋白质组成HLAⅡ类复合物,这是一种表达于抗原呈递细胞表面的α-β异质二聚体膜糖蛋白,例如B淋巴细胞、巨噬细胞和树突状细胞。HLAⅡ类糖蛋白为CD4+T细胞呈递病毒多肽从而影响免疫反应。因此HLA-DQ和-DR基因的SNPs在免疫介导的疾病中具有重要的地位,包括肝病和HCC。而WEN等[44]的近期研究发现rs9272105与HBV的基因型和变异存在显著的相关作用,并进一步揭示HLA-DQ/DR基因的多态性可能通过调节HBV变异的免疫选择而影响慢性HBV感染的结局,从而增加了因HBV变异导致HCC的风险。
3 GWAS的总结和展望
在GWAS中发现了大量HCC的易感位点,为深入研究其生物学发病机制起了指导性作用。与先前的研究方法相比,GWAS的最大优势是在研究前不需要提出致病位点所在基因组的生物学假设,而是从基因组的所有SNPs中进行筛选。采用高通量技术,辅以大样本量和重复验证,极大地提高了GWAS从海量基因组信息中筛选出与HCC相关遗传变异的效能。后续的生物学通路分析部分证实了GWAS所发现的基因的潜在作用。
但GWAS并非完美无瑕。首先,GWAS只能揭示与疾病相关的基因而不能直接验证这些基因导致疾病,因为与疾病的相关的基因与导致疾病的基因可能存在LD[45]。因此,需要功能性分析来验证GWAS所发现的HCC候选基因。其次,筛选出的位点仅能解释表型遗传的一小部分[46]。所以发现罕见变异仍然是一个问题,亟待解决[47]。另外,GWAS关注的是疾病的易感性,但并不能够反映疾病的其他方面,例如进展、严重程度或预后[48-49]。因此,有研究者在经典的GWAS基础上提出了弥补的策略和方法。随着新一代测序技术的出现和发展,GWAS将不断吸收和利用新的技术,并将其应用到将来的研究中。
[1]EL-SERAG H B.Epidemiology of viral hepatitis and hepatocellular carcinoma[J].Gastroenterology,2012,142(6): 1264-1273.
[2]FERLAY J,SHIN H R,BRAY F,et al.Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008[J].Int J Cancer,2010,127(12): 2893-2917.
[3]LAFARO K J,DEMIRJIAN A N,PAWLIK T M.Epidemiology of hepatocellular carcinoma[J].Surg Oncol Clin N Am,2015,24(1): 1-17.
[4]NAHON P,SUTTON A,ZIOL M,et al.Genetic risk markers for hepatocellular carcinoma in patients with alcoholic liver disease[J].Hepatic Oncol,2015,2(1): 63-78.
[5]BARBARA L,BENZI G,GAIANI S,et al.Natural history of small untreated hepatocellular carcinoma in cirrhosis: a multivariate analysis of prognostic factors of tumor growth rate and patient survival[J].Hepatology,1992,16(1): 132-137.
[6]KIM M N,HAN K H,AHN S H.Prevention of hepatocellular carcinoma: beyond hepatitis B vaccination[J].Semin Oncol,2015,42(2): 316-328.
[7]GOOSENS N,HOSHIDA Y.Hepatitis C virus-induced hepatocellular carcinoma[J].Clin Mol Hepatol,2015,21(2): 105-114.
[8]KEW M C.Epidemiology of hepatocellular carcinoma in sub-Saharan Africa[J].Ann Hepatol,2013,12(2): 173-182.
[9]ILES M M.What can genome-wide association studies tell us about the genetics of common disease?[J] PLoS Genet,2008,4(2): e33.
[10]MCCARTHY M I,ABECASIS G R,CARDON L R,et al.Genome-wide association studies for complex traits: consensus,uncertainty and challenges[J].Nat Rev Genet,2008,9(5): 356-369.
[11]HARDY J,SINGLETON A.Genomewide association studies and human disease[J].N Engl J Med,2009,360(17): 1759-1768.
[12]TEO Y Y.Common statistical issues in genome-wide association studies: a review on power,data quality control,genotype calling and population structure[J].Curr Opin Lipidol,2008,19(2): 133-143.
[13]CHANOCK S J,MANOLIO T,BOEHNKE M,et al.Replicating genotype-phenotype associations[J].Nature,2007,447(7145): 655-660.
[14]RISCH N,MERIKANGAS K.The future of genetic studies of complex human diseases[J].Science,1996,273(5281): 1516-1517.
[15]KLEIN R J,ZEISS C,CHEW E Y,et al.Complement factor H polymorphism in age-related macular degeneration[J].Science,2005,308(5720): 385-389.
[16]ZHANG H,ZHAI Y,HU Z,et al.Genome-wide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers[J].Nat Genet,2010,42(9): 755-758.
[17]AL-QAHTANI A,AL-ANAZI M,VISWAN N A,et al.Role of single nucleotide polymorphisms of KIF1B gene in HBV-associated viral hepatitis[J].PLoS One,2012,7(9): e45128.
[18]SAWAI H,NISHIDA N,MBAREK H,et al.No association for Chinese HBV-related hepatocellular carcinoma susceptibility SNP in other East Asian populations[J].BMC Med Genet,2012,13(1): 47.
[19]CHEN J H,WANG Y Y,LV W B,et al.Effects of interactions between environmental factors and KIF1B genetic variants on the risk of hepatocellular carcinoma in a Chinese cohort[J].World J Gastroenterol,2016,22(16): 4183-4190.
[20]BAGCHI A,MILLS A A.The quest for the 1p36 tumor suppressor[J].Cancer Res,2008,68(8): 2551-2556.
[21]LI S P,WANG H Y,LI J Q,et al.Genome-wide analyses on loss of heterozygosity in hepatocellular carcinoma in Southern China[J].J Hepatol,2001,34(6): 840-849.
[22]MIDORIKAWA Y,YAMAMOTO S,TSUJI S,et al.Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis[J].Hepatology,2009,49(2): 513-522.
[23]MUNIRAJANA K,ANDO K,MUKAI A,et al.KIF1Bbeta functions as a haploinsufficient tumor suppressor gene mapped to chromosome 1p36.2 by inducing apoptotic cell death[J].J Biol Chem,2008,283(36): 24426-24434.
[24]SCHILISIOS,KENCHAPPA R S,VREDEVELD L C,et al.The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor[J].Genes Dev,2008,22(7): 884-893.
[25]MIKI D,OCHI H,HAYES C N,et al.Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers[J].Nat Genet,2011,43(8): 797-800.
[26]HARADA Y,KANEHIRA M,FUJISAWA Y,et al.Cell-permeable peptide DEPDC1-ZNF224 interferes with transcriptional repression and oncogenicity in bladder cancer cells[J].Cancer Res,2010,70(14): 5829-5839.
[27]BURZA M A,MOTTA B M,MANCINA R M,et al.DEPDC5 variants increase fibrosis progression in Europeans with chronic hepatitis C virus infection[J].Hepatology,2016,63(2): 418-427.
[28]SANGIOVANNI A,PRATI G M,FASANI P,et al.The natural history of compensated cirrhosis due to hepatitis C virus:a 17-year cohort study of 214 patients[J].Hepatology,2006,43(6): 1303-1310.
[29]KUMAR V,KATO N,URABE Y,et al.Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma[J].Nat Genet,2011,43(5): 455-458.
[30]VIVIER E,TOMASELLO E,BARATIN M,et al.Functions of natural killer cells[J].Nat Immunol,2008,9(5): 503-510.
[31]WANG H,YANG D,XU W,et al.Tumor-derived soluble MICs impair CD3(+)CD56(+) NKT-like cell cytotoxicity in cancer patients[J].Immunol Lett,2008,120(1-2): 65-71.
[32]JIANG X,ZOU Y,HUO Z,et al.Association of major histocompatibility complex class Ⅰ chain-related gene A microsatellite polymorphism and hepatocellular carcinoma in South China Han population[J].Tissue Antigens,2011,78(2): 143-147.
[33]TATSUMI T,TAKEHARA T.Impact of natural killer cells on chronic hepatitis C and hepatocellular carcinoma[J].Hepatol Res,2016,46(5): 416-422.
[34]LI S,QIAN J,YANG Y,et al.GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers[J].PLoS Genet,2012,8(7): e1002791.
[35]DE GROOT J,SONTHEIMER H.Glutamate and the biology of gliomas[J].Glia,2011,59(8): 1181-1189.
[36]SPEYER C L,SMITH J S,BANDA M,et al.Metabotropic glutamate receptor-1: a potential therapeutic target for the treatment of breast cancer[J].Breast Cancer Res Treat,2012,132(2): 565-573.
[37]STEPULAK A,LUKSCH H,UCKERMANN O,et al.Glutamate receptors in laryngeal cancer cells[J].Anticancer Res,2011,31(2): 565-573.
[38]HERNERr A,SAULIUNAITE D,MICHALSKI C W,et al.Glutamate increases pancreatic cancer cell invasion and migration via AMPA receptor activation and Kras-MAPK signaling[J].Int J Cancer,2011,129(10): 2349-2359.
[39]JIANG D K,SUN J,CAO G,et al.Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma[J].Nat Genet,2013,45(1): 72-75.
[40]YU H,JOVE R.The STATs of cancer-new molecular targets come of age[J].Nat Rev Cancer,2004,4(2): 97-105.
[41]LIAO Y,CAI B,LI Y,et al.Association of HLA-DP/DQ and STAT4 polymorphisms with HBV infection outcomes and a mini meta-analysis[J].PLoS One,2014,9(11): e111677.
[42]WEAVER J R,NADLERR J L,TAYLOR-FISHWICK D A.Interleukin-12 (IL-12)/STAT4 axis is an important element for β-cell dysfunction induced by inflammatory cytokines[J].PLoS One,2015,10(11): e0142735.
[43]HORRAS C J,LAMB C L,MITCHELL K A.Regulation of hepatocyte fate by interferon-gamma[J].Cytokine Growth Factor Rev,2011,22(1): 35-43.
[44]WEN J,SONG C,JIANG D,et al.Hepatitis B virus genotype,mutations,human leukocyte antigen polymorphisms and their interactions in hepatocellular carcinoma: a multi-centre case-control study[J].Sci Rep,2015,5: 16489.
[45]MARIAN A J.Molecular genetic studies of complex phenotypes[J].Transl Res,2012,159(2): 64-79.
[46]MANOLIO T A,COLLINS F S,COX N J,et al.Finding the missing heritability of complex diseases[J].Nature,2009,461(7265): 747-753.
[47]MCCLELLAN J,KING M C.Genetic heterogeneity in human disease[J].Cell,2010,141(2): 210-217.
[48]CHAN K Y,WONG C M,KWAN J S,et al.Genome-wide association study of hepatocellular carcinoma in Southern Chinese patients with chronic hepatitis B virus infection[J].PLoS One,2011,6(12): e28798.
[49]QU L S,JIN F E I,GUO Y M,et al.Nine susceptibility loci for hepatitis B virus-related hepatocellular carcinoma identified by a pilot two-stage genome-wide association study[J].Oncology Letters,2016,11(1): 624-632.
猜你喜欢
区域治理(2022年40期)2022-11-27
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
中成药(2018年7期)2018-08-04
百科知识(2015年18期)2015-09-10
