
具有氧空位BixWO6(1.81≤x≤2.01)的第一性原理计算和光催化性能研究∗
2018-03-26 19:06:52何金云彭代江王燕舞龙飞邹正光
物理学报 2018年6期
何金云 彭代江 王燕舞 龙飞 邹正光
1)(桂林理工大学,有色金属及材料加工新技术教育部重点实验室,桂林 541004)
2)(桂林理工大学,广西建筑新能源与节能重点实验室,桂林 541004)
3)(桂林理工大学环境科学与工程学院,桂林 541004)
1 引 言
自1972年Fujishima和Honda[1]发现可以通过在TiO2电极上分解水产生氢气以来,光催化过程形成的人工光合作用被认为是解决当前的环境和能源问题的希望.实验证明在氧化物半导体和非氧化物半导体中TiO2是最合适的光催化材料[2],同时具有低成本、环境友好、化学稳定性好且易于制备.但是TiO2有较宽的带隙,锐钛矿相3.2 eV,金红石相3.0 eV,它只能吸收太阳光谱中不到5%的紫外光,而约50%的可见光无法吸收,因此实际应用范围受限.
主要有两种方法开发新的光催化剂,一种是对TiO2采用助催化剂进行表面修饰或者通过掺杂来扩展它的吸收边到可见光区,以提高它对太阳能的利用率[3−6];另一种方法是寻找带宽合适、吸收范围在可见光区的新型光催化材料[7].近年来人们发现Bi基氧化物具有可见光吸收带,价带顶主要是由Bi 6s和O 2p杂化形成的填满的反键态,从价带顶到导带的跃迁所需能量部分位于可见光区[8−11].Bi基氧化物具有无毒、化学和热力学稳定[12].在Bi基氧化物中,Bi2WO6的禁带宽度为2.7 eV,能被紫外光和可见光激发.同时,Bi2WO6合成过程中形貌可控、氧化能力强、耐光腐蚀[13−16],作为新型可见光光催化剂应用前景广阔.
提高Bi2WO6的光催化活性,抑制光生载流子的复合是很重要的.氧空位可作为电子俘获中心,因而它在抑制光生载流子复合方面具有重要的作用[17−19].而且氧空位还可作为反应物分子的反应中心[20].Zhang等[21]合成了具有氧空位的锆离子掺杂的Bi2WO6,其光催化性能提高了3倍.Nie等[22]合成了具有氧空位的纳米凹片状Bi2WO6,它具有很高的电催化产氧性能.因此,将氧空位引入到光催化材料中,有望获得高活性的光催化材料.
本文采用溶剂热法合成了具有氧空位的BixWO6(1.81≤x≤2.01).采用密度泛函理论(DFT)计算和实验系统研究了Bi含量和氧空位对Bi2WO6的电子结构、晶体结构和光催化性能的影响.
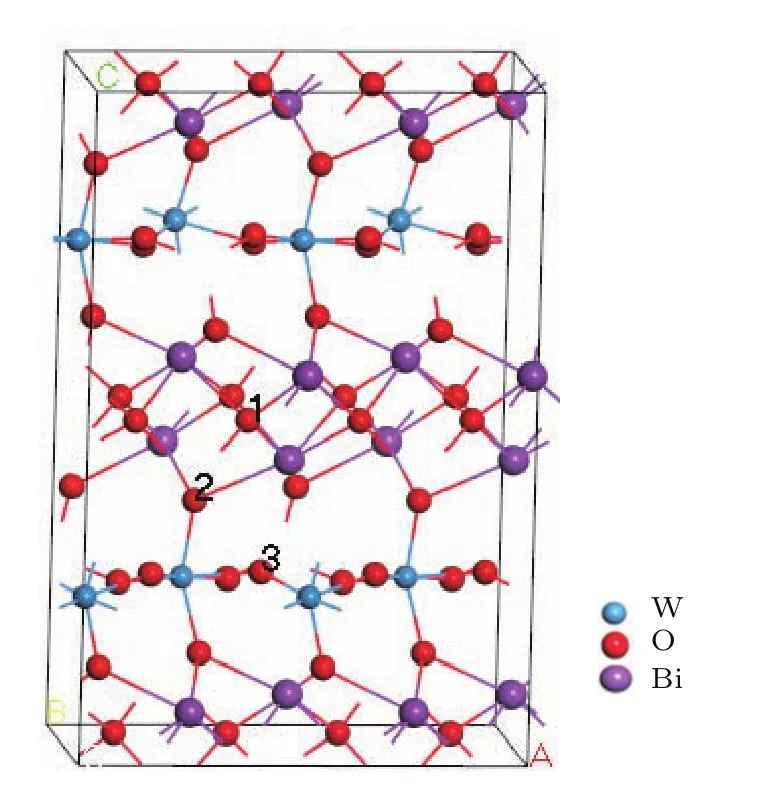
图1 包含72个原子的Bi2WO6晶胞和三种氧缺陷的位置Fig.1.Bi2WO6cell with 72 atoms and three kinds of oxygen vacancies.
2 实 验
2.1 DFT理论计算方法和模型
采用第一性原理计算了氧空位对Bi2WO6晶体结构和电子结构的影响. 采用创腾公司Material Studio软件中的Castep模块.计算模型采用包含72个原子的2×1×1 Bi2WO6超胞,原胞晶格参数采用McDowell的实验值:a=5.488 Å,b=5.4607 Å,c=16.4842 Å, 空 间组群为PCA21[23],模型结构如图1所示.采用Broyden-Fletcher-Goldfarb-Shanno优化算法;交换-关联势采用广义梯度近似中的PBESOL泛函[24]来处理电子与电子之间的相互作用交换相关能,对Bi,O,W加超软赝势来描述价电子与离子实之间的相互作用,计算时分别将Bi的6s26p3,O的2s22p4,W的5d46s2轨道电子作为价电子,电子波函数用平面波基组展开,平面波截断能经过收敛测试后设为380 eV,体系的总能量和电荷密度计算在K空间布里渊区积分通过Monkhorst-Pack方法产生的3×6×2点网络下进行,保证了体系能量和构型在准完备平面波基组水平上的收敛.对几何结构进行优化的收敛标准为:体系总能量变化≤5×10−6eV/atom,原子受力≤0.05 eV/Å,原子间内应力≤0.1 GPa.在自洽场运算中,电子最小化采用密度混合法,密度混合采用了Pulay混合法,自洽场收敛精度为5×10−6eV/atom.能带计算时在K空间沿以下高对称点进行:G(0,0,0),F(0,0.5,0),Q(0,0.5,0.5)和Z(0,0,0.5).原子和成键布居分析采用Mulliken布居分析.如图1所示,在Bi2WO6超胞中分别有三种O缺陷位,分别标记为O1,O2,O3,计算过程中固定晶胞晶格参数,只优化原子位置.
2.2 具有氧空位BixWO6(1.81≤x≤2.01)光催化剂的合成
采用溶剂热法合成具有氧空位的非化学计量BixWO6(x=1.81,1.87,1.89,1.92,2.01)光催化剂.具体过程如下:将10 mmol Na2WO4·5 H2O溶解于30 mL去离子水中.同时,将不同质量的Bi(NO3)3·3H2O(18.5,19,19.5,20和20.5 mmol)80°C溶解于30 mL乙酸中,将这两种溶液混合后搅拌1 h.然后将这五组混合液倒入100 mL水热釜中,放入烘箱,于180°C反应15 h.自然冷却到室温后,分别将所得到的沉淀物用去离子水和无水乙醇离心洗涤3次,于80°C烘干,得到BixWO6(x=1.81,1.87,1.89,1.92,2.01)产品,并标记为BWO-X(X=1,2,3,4,5).
3 结果与讨论
3.1 DFT计算
3.1.1 Bi2WO6−y的氧空位缺陷形成能
缺陷形成能是衡量一个体系是否容易形成以及结构相对稳定性的重要指标,根据下式计算缺陷形成能Ef:
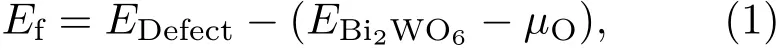
其中,Ef为形成能,Edefect为缺陷体系的能量,EBi2WO6为未掺杂Bi2WO6体系的能量,µO为O原子的化学势,计算结果如表1所列.

表1 具有一个氧空位缺陷的Bi16W8O48的形成能Table 1.Formation energy of Bi16W8O48with one oxygen vacancy.
形成能越小,说明缺陷越容易形成,反之则越不容易形成.由表1可知O1位缺陷最有可能形成,而O3缺陷的形成难度最大.
3.1.2 氧空位缺陷态Bi2WO6−y的原子间键长和布居分析
原子间键长和电荷布居的计算结果表明,氧空位缺陷主要影响与之有化学键作用的外两层电子,因此主要选择分析与Bi原子相邻的6个O原子位置及与其成键的Bi,W原子,氧空位缺陷形成前后相应的原子间键长如图2.
从图2可以看出:形成O1位缺陷后,主要影响[Bi2O2]2+层,但总体影响不大,晶体内部其他原子间键长变化也不明显,晶格畸变较小;形成O2位缺陷后,主要影响[Bi2O2]2+和[WO6]6−层间,但总体影响不大,只引起缺陷周围W—O,Bi—O之间键长轻微减小,晶体内部其他原子间键长变化不明显,晶格畸变较小;形成O3位缺陷后,主要影响[WO6]6−层内的W,O原子,只引起缺陷周围W—O键长轻微减小,晶体内部其他原子间键长变化不明显,晶格畸变较小.因此,三种位置的氧缺陷引起的晶格畸变都较小,对Bi2WO6的晶体结构无明显影响.
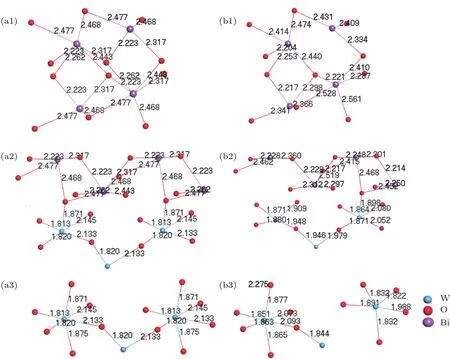
图2 无缺陷及有一个氧空位缺陷Bi16W8O48晶胞的键长(单位Å) (a1)无缺陷O1位;(b1)缺陷O1位;(a2)无缺陷O2位;(b2)缺陷O2位;(a3)无缺陷O3位;(b3)缺陷O3位Fig.2.Bond length of Bi16W8O48cell with or without one oxygen vacancy(unit:Å):(a1)Without O1 oxygen vacancy;(b1)with O1 oxygen vacancy;(a2)without O2 oxygen vacancy;(b2)with O2 oxygen vacancy;(a3)without O3 oxygen vacancy;(b3)with O3 oxygen vacancy.
图3和表2直观地显示了形成氧空位缺陷后原子电荷布居的变化.可以看出,O1空位形成后,缺陷位相邻Bi原子电荷布居明显减小,氧原子电荷基本保持不变,Bi—O键布居减小,共价性减弱,主要是由于氧原子电负性较强,形成空位缺陷后周围Bi原子所需提供电子数相应减小;其他Bi—O键影响不大,共价性略微增强.形成O2空位后,缺陷周围Bi,W原子电荷布居均减小,但W原子周围的氧原子电荷数增加,总体上键长减小、键布居增加,原子间共价性作用增强.形成O3位缺陷后,W原子电荷布居减小,主要是由于减少了一个W—O键,缺陷位相邻W原子周围的氧原子电荷数增加,W—O键布居增加,共价性增强,总体上导致W—O间作用增强.计算结果说明,氧空位使Bi2WO6的晶体结构生成了更多的缺陷,一定数目的晶体缺陷可有效俘获光生电子,抑制电子-空穴对的复合,从而提高半导体的光催化活性.

图3 无缺陷及有一个氧空位缺陷的Bi16W8O48晶胞电荷布居 (a1)无缺陷O1位;(b1)缺陷O1位;(a2)无缺陷O2位;(b2)缺陷O2位;(a3)无缺陷O3位;(b3)缺陷O3位Fig.3.Charge population of Bi16W8O48cell with or without one oxygen vacancy:(a1)Without O1 oxygen vacancy;(b1)with O1 oxygen vacancy;(a2)without O2 oxygen vacancy;(b2)with O2 oxygen vacancy;(a3)without O3 oxygen vacancy;(b3)with O3 oxygen vacancy.
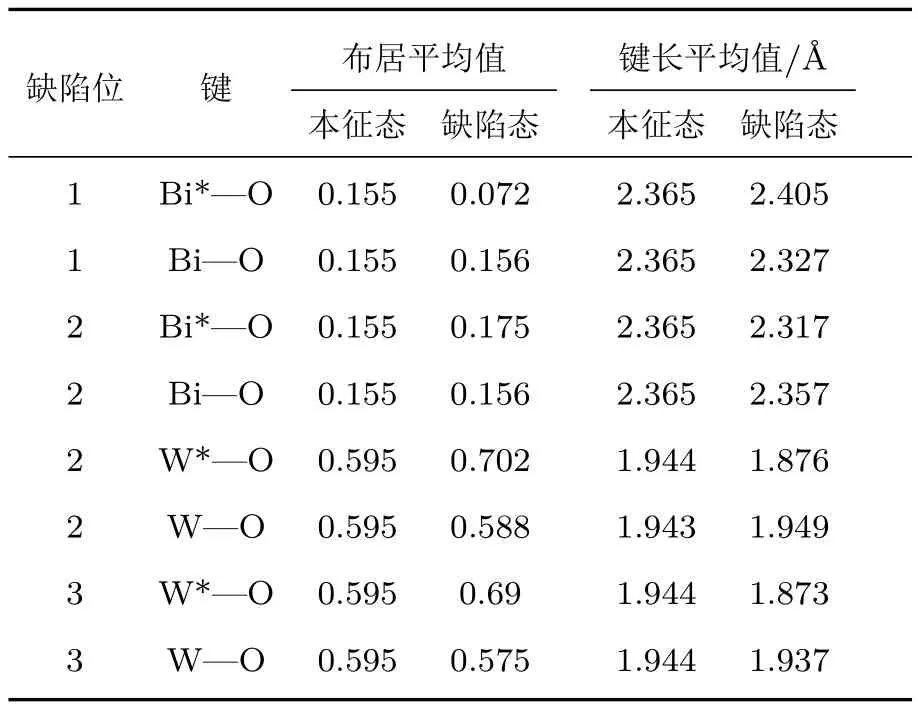
表2 氧空位附近的原子键布居和键长Table 2.Bond population and bond length of atom near oxygen vacancy.
3.1.3 氧空位缺陷态Bi2WO6−y的电子结构分析
由图4形成氧空位缺陷后的能带结构及图5总态密度图可以看出,三种氧空位均导致体系禁带宽度变窄,导带底和价带顶能量均降低,与文献[25]的实验结果一致.其中O1位带隙降低是由于禁带中间形成了杂质能级,主要是Bi的6s轨道和O的2p轨道杂化引起,可能是因为Bi—O间电子相互作用增强所致.O2,O3位带隙降低是由于W—O间电子相互作用增强导致导带底能级离散,同时W的5d和O的2p杂化轨道能量降低引起.由于形成空位后费米能级均位于导带底,因此都是n型半导体.由此可见,三种氧空位都会导致体系带隙变窄,使光生电子跃迁更加容易,有利于提高Bi2WO6的光催化性能.由此,我们采用溶剂热法合成了具有氧空位的BixWO6(1.81≤x≤2.01)产品,以获得高活性可见光催化材料.
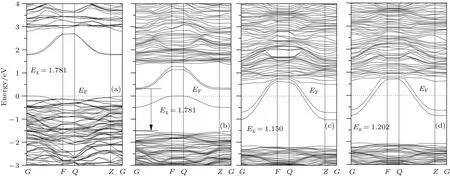
图4 本征态及形成氧空位缺陷后的Bi16W8O48晶胞能带结构图 (a)本征态;(b)O1位空位;(c)O2位空位;(d)O3位空位Fig.4.Calculated band structure of Bi16W8O48cell:(a)Eigenstate;(b)oxygen vacancy at O1;(c)oxygen vacancy at O2;(d)oxygen vacancy at O3.

图5 本征态及形成O空位缺陷后的Bi16W8O48晶胞总态密度图 (a)本征态;(b)O1位空位;(c)O2位空位;(d)O3位空位Fig.5.Total density of states of Bi16W8O48cell:(a)Eigenstate;(b)oxygen vacancy at O1;(c)oxygen vacancy at O2;(d)oxygen vacancy at O3.
3.1.4 氧空位缺陷态Bi2WO6−y的吸收光谱分析
从晶胞吸收光谱图(图6)可看出,形成氧空位缺陷后,晶体在可见光区吸收强度都有所增加,其中O1空位情况下增加较多.原因主要是形成氧空位缺陷后,体系的带隙减小.O1空位可见光区吸收强度增加较多的原因是O1空位还形成了杂质能级.结果表明,具有氧空位的半导体的光催化活性较没有氧空位的高.
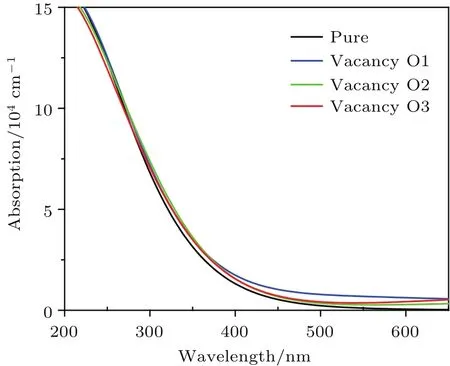
图6 本征态及形成氧空位缺陷后Bi16W8O48晶胞的吸收光谱图Fig.6.Absorption spectra of eigen Bi16W8O48cell and Bi16W8O48cell with oxygen vacancy defects.

图7 BixWO6产品的X射线衍射谱图 (a)全谱图;(b)放大图Fig.7. XRD patterns of the BixWO6products:(a)Full spectra,(b)enlarged patterns.

表3 BixWO6产品的XRF结果Table 3.XRF results of the BixWO6products.
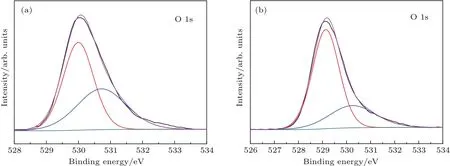
图8 (a)BWO-3和(b)BWO-5产品的XPS高分辨O 1s图谱Fig.8.High-resolution XPS spectra of O 1s for(a)BWO-3 and(b)BWO-5 products.
3.2 BixWO6(1.81≤x≤2.01)的实验研究
3.2.1 BixWO6(1.81≤x≤2.01)产品的相组成
图7为BWO-X(X=1,2,3,4,5)产品的X射线衍射(XRD)谱.从图7可以看出,所有产品的XRD衍射峰都属于正交相Bi2WO6(JCPDS No.39-0256),没有发现其他物相的衍射峰,说明所有产品都是纯的Bi2WO6产品.表3为产品的X射线荧光光谱(XRF)分析数据,可以看出,所有产品的Bi,W摩尔比都较它们的理论值低.BWO-5产品的Bi,W摩尔为2.01,与Bi2WO6的理论化学计量比相近.
采用X射线光电子能谱(XPS)研究了BWO-3和BWO-5产品氧空位的存在情况(图8). 从BWO-3和BWO-5产品的O 1s谱可以看出,它们都由一个低结合能峰和一个高结合能峰组成,这两个结合能峰分别归属于晶格氧和化学吸附氧,如羟基和H2O[26],其中高结合能O 1s峰的面积随着氧空位的增加而增加[27].BWO-3和BWO-5产品的高结合能O 1s峰的面积比为1.18,说明BWO-3中存在较多的氧空位.由DFT计算结果可知,氧空位会导致Bi2WO6的带隙变窄,使光电子跃迁更加容易,因而BWO-3产品具有更好的光催化性能.
3.2.2 合成BixWO6(1.81≤x≤2.01)产品的显微结构
产品的形貌和微观结构通过场发射扫描电子显微镜(FESEM)和透射电子显微镜(TEM)分析得到.从BWO-X(X=1,2,3,4,5)产品的FESEM图可以看出,所有产品的形貌都由花状微球和纳米片组成,花状微球的平均粒径为2—3µm(图9).从BWO-3产品的放大图可以看出,这些微球由一些纳米片构成.从BWO-3样品的TEM图可知,这些纳米片由尺寸小于100 nm的纳米薄片组装而成(图10(a)).这些纳米薄片上的HRTEM图可看到清晰的宽度为0.27 nm的晶格条纹(图10(b)),归属于Bi2WO6的(006)晶面.
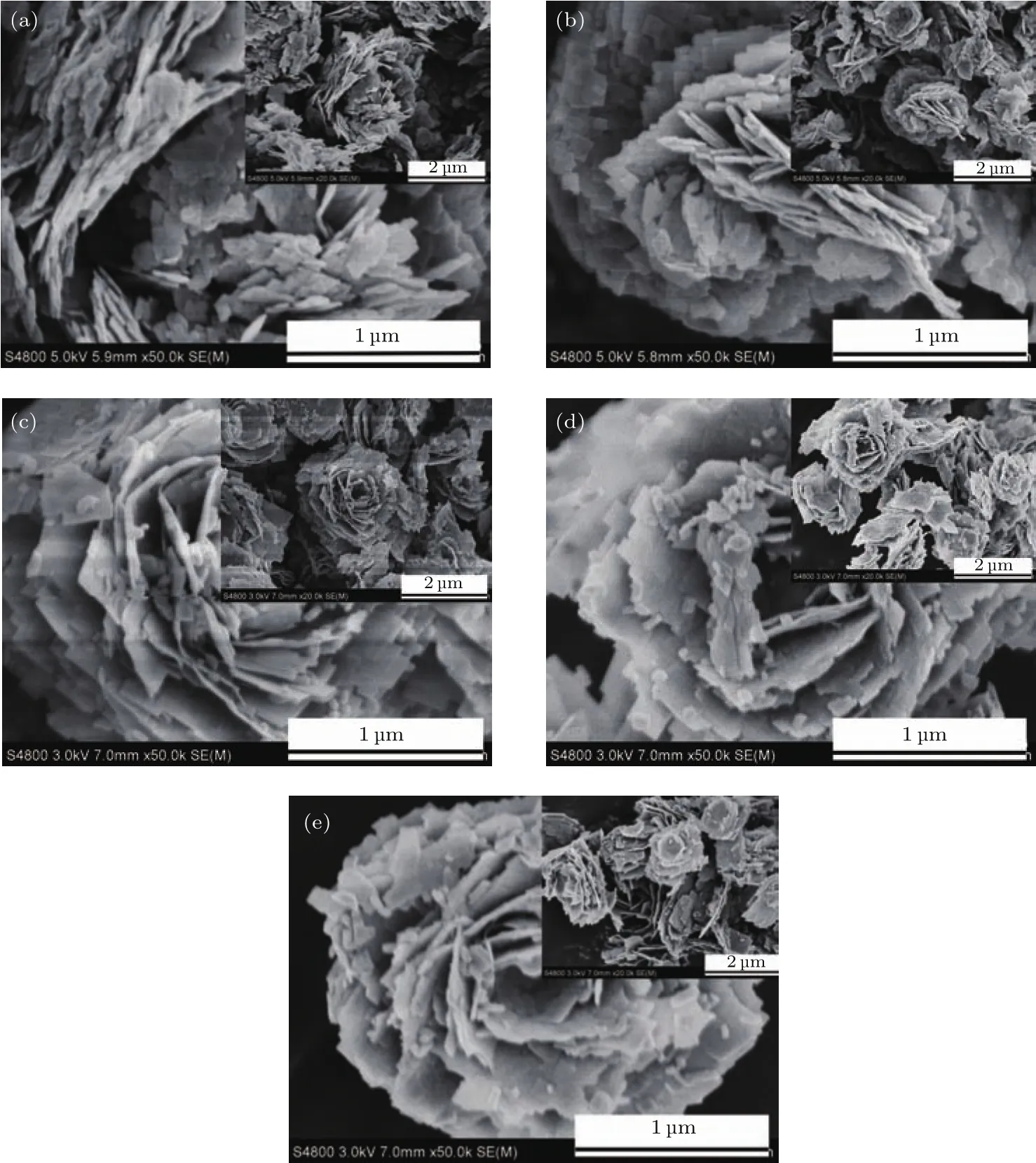
图9 产品的FE-SEM图像 (a)BWO-1;(b)BWO-2;(c)BWO-3;(d)BWO-4;(e)BWO-5Fig.9.FE-SEM images of the products:(a)BWO-1;(b)BWO-2;(c)BWO-3;(d)BWO-4;(e)BWO-5.

图10BWO-3产品的(a)TEM和(b)HRTEM图像Fig.10.(a)TEM and(b)HRTEM image of the BWO-3 product.

图11 (a)样品的紫外-可见吸收光谱;(b)样品可见光催化降解RhB溶液(1×10–5mol/L)的性能;(c)以BWO-3产品为催化剂,可见光(λ>420 nm)照射下,RhB水溶液随光照时间变化的吸收光谱;(d)BWO-3和BWO-5产品的荧光光谱Fig.11.(a)UV-visible diffuse re fl ectance spectra of the samples;(b)UV-visible spectral changes of RhB(1×10−5 mol/L)in aqueous catalyst dispersions as a function of irradiation time;(c)the temporal evolution of the spectrum during the photodegradation of RhB mediated by BWO-3 catalyst under visible light illumination(λ> 420 nm);(d)PL spectra of BWO-3 and BWO-5 product.
3.2.3 紫外可见光吸收谱和光催化性能
影响半导体光催化性能的另一关键因素是其能带结构[28].图11(a)为产品的紫外-可见光吸收谱.从图11(a)可以看出,所有样品的光吸收边都从紫外区延伸到460 nm处的可见光区,说明这些样品都具有可见光催化活性,实验结果与图6的DFT计算结果相一致.在这些产品中,BWO-3产品在可见光区的吸收最强,预示了其最好的光催化性能.
为了表征BWO-X(X=1,2,3,4,5)产品的光催化性能,研究了可见光(λ>420 nm)照射下样品催化降解罗丹明B(RhB)水溶液的性能.图11(b)为以BWO-X(X=1,2,3,4,5)为催化剂时,不同光照时间RhB的降解率.C0和C分别为光照前和光照一定时间后RhB的浓度.在同等降解实验条件下,以未放催化剂的空白实验和TiO2(P-25)催化剂作为对比实验.从图11(b)可以看出,光照180 min后,未放催化剂的空白实验中,RhB几乎未发生降解,说明RhB的光稳定性很好.而使用P-25为催化剂时,RhB只降解了8%.而本文的样品中,BWO-3样品的光催化性能最好,RhB降解了98%.BWO-4样品的光催化性能略差于BWO-3样品.而BWO-2和BWO-5样品由于其Bi,W比相差不大,因此它们的光催化性能也相差不大,RhB都降解了96%.当使用BWO-3为催化剂,光照时间不同时,RhB溶液的光吸收谱的变化如图11(c)所示.RhB主要的特征吸收峰位于553 nm波长处,光照射180 min后,RhB几乎被完全降解,其乙基基团被氧化脱除,染料的颜色也由玫瑰红变为无色.
由于荧光发射主要来源于半导体中自由载流子的复合,因而荧光发射光谱常被用来表征载流子复合率的高低.材料的光生电子和空穴的复合速率越高,其荧光激发光谱强度越高.光生电子和空穴的复合率是影响光催化活性的主要因素,复合率越小,光催化活性越高.图11(d)为BWO-3和BWO-5的荧光光谱图,BWO-3的荧光强度较BWO-5的弱,这与其光催化性能较好是相一致的.这是因为,非化学计量导致BWO-3产品中存在较多的氧空位,生成带正电荷的VO缺陷,它们可作为俘获中心俘获光生电子,减少光生电子和空穴对的复合[29].
从以上DFT计算、实验研究的结果分析可知,BWO-3产品光催化性能最好的原因如下:一方面,非化学计量导致BWO-3产品中存在较多的氧空位,生成带正电荷的VO缺陷,它们可作为俘获中心俘获光生电子,减少光生电子和空穴对的复合[29];元素的非化学计量也可以促进光生电子的产生,俘获水分子,生成更多的O2−[30].由DFT计算可知,产生氧空位后,氧空位缺陷导致Bi2WO6的带隙变窄,使光电子跃迁更加容易;同时,氧空位使Bi2WO6的晶体结构生成更多的缺陷,一定数目的晶体缺陷可有效俘获光生电子,抑制电子-空穴对的复合.因此,Bi元素的非化学计量和氧空位的协同作用使BWO-3产品的光催化性能最好.
4 结 论
采用溶剂热法合成了具有氧空位的非化学计量BixWO6(x=1.81,1.87,1.89,1.92,2.01)光催化剂.DFT计算结果表明,氧空位的存在可显著减小Bi2WO6的带隙,有利于光生电子的生成,并有效抑制电子空穴的复合.实验结果表明,Bi元素的含量小于化学计量比时,Bi2WO6的晶体结构发生了微小变形,Bi元素含量对Bi2WO6的显微结构影响不大,但会影响产品氧空位的含量和光吸收性能.Bi1.89WO6产品的光催化性能最佳,可见光照射180 min后,可降解98%的RhB.采用适当方法,使产品具有氧空位和非化学计量是获得高光催化活性材料的一种有效方法.
[1]Fujishima A,Honda K 1972Nature238 37
[2]Jing L,Sun X 2003Sol.Energy Mater.Sol.Cells79 133
[3]Liu Y,Yu L,Wei Z G,Pan Z C,Zou Y D,Xie Y H 2013Chem.J.Chin.Univ.(in Chinese)[刘月,余林,魏志钢,潘湛昌,邹燕娣,谢英豪2013高等学校化学学报34 434]
[4]Carp O,Huisman C L,Reller A 2004Prog.Solid State Chem.32 33
[5]Yang K,Dai Y,Huang B 2008Chem.Phys.Lett.456 71
[6]Wang P,Huang B,Lou Z 2010Chem.Eur.J.16 538
[7]Kubacka A,Fern Ndezgarc A M,Col N G 2012Chem.Rev.112 1555
[8]Kudo A,Omori K,Kato H 1999J.Am.Chem.Soc.121 11459
[9]Fu H,Pan C,Yao W 2005J.Phys.Chem.B109 22432
[10]Zhang L,Wang W,Yang 2006J.Appl.Catal.A308 105
[11]Lai K,Zhu Y,Lu J 2013Comput.Mater.Sci.67 88
[12]Zeng D W,Xie C S,Zhu B L 2003Mater.Sci.Eng.B104 68
[13]Zhang L,Wang W,Zhou L 2007Small3 1618
[14]Zhang Z,Wang W,Gao E 2012J.Phys.Chem.C116 25898
[15]Bhattacharya C,Lee H C,Bard A J 2013J.Phys.Chem.C117 9633
[16]Sun Z X,Li X F,Guo S,Wang H Q,Wu Z B 2013J.Colloid Interf.Sci.412 31
[17]Kuo T J,Lin C N,Kuo C L,Huang M H 2007Chem.Mater.19 5143
[18]Wang J C,Liu P,Fu X Z,Li Z H,Han W,Wang X X 2009Langmuir.25 1218
[19]Zheng Y H,Chen C Q,Zhan Y Y,Lin X Y,Zheng Q,Wei K M,Zhu J F,Zhu Y J 2007Inorg.Chem.46 6675
[20]Gong X Q,Selloni A,Batzil M 2006Nat.Mater.5 665
[21]Zhang Z,Wang W,Gao E,Shang M,Xu J 2011J.Hazard Mater.196 255
[22]Nie Z,Ma D,Fang G Y,Chen W,Huang S M 2016J.Mater.Chem.A4 2438
[23]Mcdowell N A,Knight K S 2006Chem.Eur.J.12 1493
[24]Perdew J P,Ruzsinszky A,Csonka G I 2008Phys.Rev.Lett.101 136406
[25]Lu Q,Hua L G,Chen Y L 2015J.Inorg.Mater.30 413(in Chinese)[卢青,华罗光,陈亦琳2015无机材料学报30 413]
[26]Zhou B,Zhao X,Liu H 2010Appl.Catal.B99 214
[27]Sun S B,Chang X T,Li Z J 2012Mater.Charact.73 130
[28]Lin Z,Wang W,Liu S 2006J.Mol.Catal.A252 120
[29]Wu J,Duan F,Zheng Y 2007J.Phys.Chem.C111 12866
[30]Ding X,Zhao K,Zhang L 2014Environ.Sci.Technol.48 5823
猜你喜欢
高中数理化(2024年4期)2024-03-16 11:09:37
高中数理化(2022年16期)2022-09-14 13:57:04
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08 03:23:36
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
陶瓷学报(2019年5期)2019-01-12 09:17:38
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
读者欣赏(2014年6期)2014-07-03 03:00:48
语文知识(2014年2期)2014-02-28 21:59:21
