
代谢工程改造大肠杆菌提高乙醇酸产率
2018-03-23 05:27马宁朱康佳毛银邓禹
生物工程学报 2018年2期
马宁,朱康佳,毛银,邓禹
江南大学 粮食发酵工艺与技术国家工程实验室,江苏 无锡 214122
乙醇酸 (Glycolic acid, Glycolate) 又称羟基乙酸、甘醇酸,是最简单的α-羟基酸[1]。近年来,乙醇酸广泛应用于纺织工业、医学工程材料、化学清洗等许多领域,需求量逐年增加[2-3]。
已报道的生物合成乙醇酸的方法包括微生物酶催化法和全生物合成方法。主要的生物酶催化法包括利用产腈水解酶水解乙醇腈产乙醇酸[4],产甘油氧化酶氧化乙二醇产乙醇酸[2],以及利用化学合金铁硫氧化细菌合成乙醇酸[5]。但是,上述生产乙醇酸的方法需要昂贵或有毒的前体物质,使得生物催化法生产乙醇酸的应用受到限制。为了解决上述问题,研究人员尝试利用葡萄糖或木糖作为碳源,通过过量表达乙醇酸合成途径中3个关键酶:异柠檬酸裂解酶 (aceA)、乙醛酸还原酶 (ycdW) 和异柠檬酸脱氢酶激酶/磷酸化酶 (aceK),加强乙醛酸循环途径,实现乙醇酸的积累[1,6-8]。Pereira等[9]以木糖为碳源合成乙醇酸,在大肠杆菌中构建木糖代谢途径并加强乙醛酸循环,获得乙醇酸产率为0.63 g/g 木糖 (理论产率的 63%),在目前所报道的利用木糖生产乙醇酸的研究中,产率达到最高。但以木糖为碳源生产乙醇酸,原料成本高昂,难以满足工业生产的需要。Dischert等[10]在大肠杆菌中以葡萄糖为底物,通过加强乙醛酸循环实现了乙醇酸的积累,产率为 0.132 g/g葡萄糖,低于理论产率(0.85 g/g葡萄糖),所以本研究的目的在于有效提高乙醇酸的产率。
乙醇酸的生物合成途径包括 3个关键酶 (异柠檬酸裂解酶aceA、乙醛酸还原酶ycdW和异柠檬酸脱氢酶激酶/磷酸化酶aceK) 和2个关键反应(异柠檬酸裂解生成乙醛酸和乙醛酸转化为乙醇酸),因此,如何调节3个关键酶的表达水平和两步反应的反应速率是提高乙醇酸产率的关键。在本研究中首先利用不同启动子调节关键基因(aceA、ycdW和acek) 的表达水平,实现各基因间协调表达;然后过量表达了柠檬酸合成酶(gltA),进一步加强乙醛酸循环;敲除了乙醇酸合成的竞争途径,阻断乙醇酸合成前体乙醛酸的消耗。最终获得了工程菌株 Mgly335,其乙醇酸产率达到0.522 g/g葡萄糖 (理论产率的61.4%)。
1 材料与方法
1.1 菌种和质粒
实验所用菌株及质粒见表1。
1.2 培养基及培养方法
实验所用培养基包括 LB培养基 (含 10 g/L胰蛋白胨,5 g/L酵母粉,10 g/L氯化钠) 和以葡萄糖为唯一碳源的 M9培养基 (6.78 g/L Na2HPO4、3 g/L KH2PO4、1 g/L NH4Cl、0.5 g/L NaCl、2 mmol/L MgSO4、0.1 mmol/L CaCl2) 并加入相应的抗生素 (50 mg/L卡那霉素,50 mg/L链霉素,100 mg/L氨苄青霉素)。
培养方法:将–80 ℃保藏菌株接种在添加相应抗生素的 LB固体平板上分离活化后,转接至50 mL LB液体培养基中,37 ℃、200 r/min培养过夜,获得摇瓶发酵种子液。取 1 mL种子液接种于50 mL M9培养基中,37 ℃、200 r/min培养至对数中期 (OD600=0.6-0.8),加入相应诱导剂(1 mmol/L IPTG或250 μg/L 脱水四环素) 以诱导目的基因表达。每4-6 h取一次样,用HPLC检测乙醇酸产量。所有的摇瓶发酵设置3个平行,最终结果为3个平行的平均值。
分批发酵培养方法:甘油保藏的菌种于平板上划线活化后,挑取单菌落接种于50 mL的LB液体培养基中,37 ℃、200 r/min摇瓶过夜。以1%接种量将活化后的菌种转接于60 mL LB液体培养基中,37 ℃、200 r/min培养6 h作为种子备用,以2%的接种量接种于5 L发酵罐中 (装液量为3 L)。发酵条件如下:搅拌转速400 r/min,通气量1 vvm,2 mol/L NaOH维持pH为7.0。
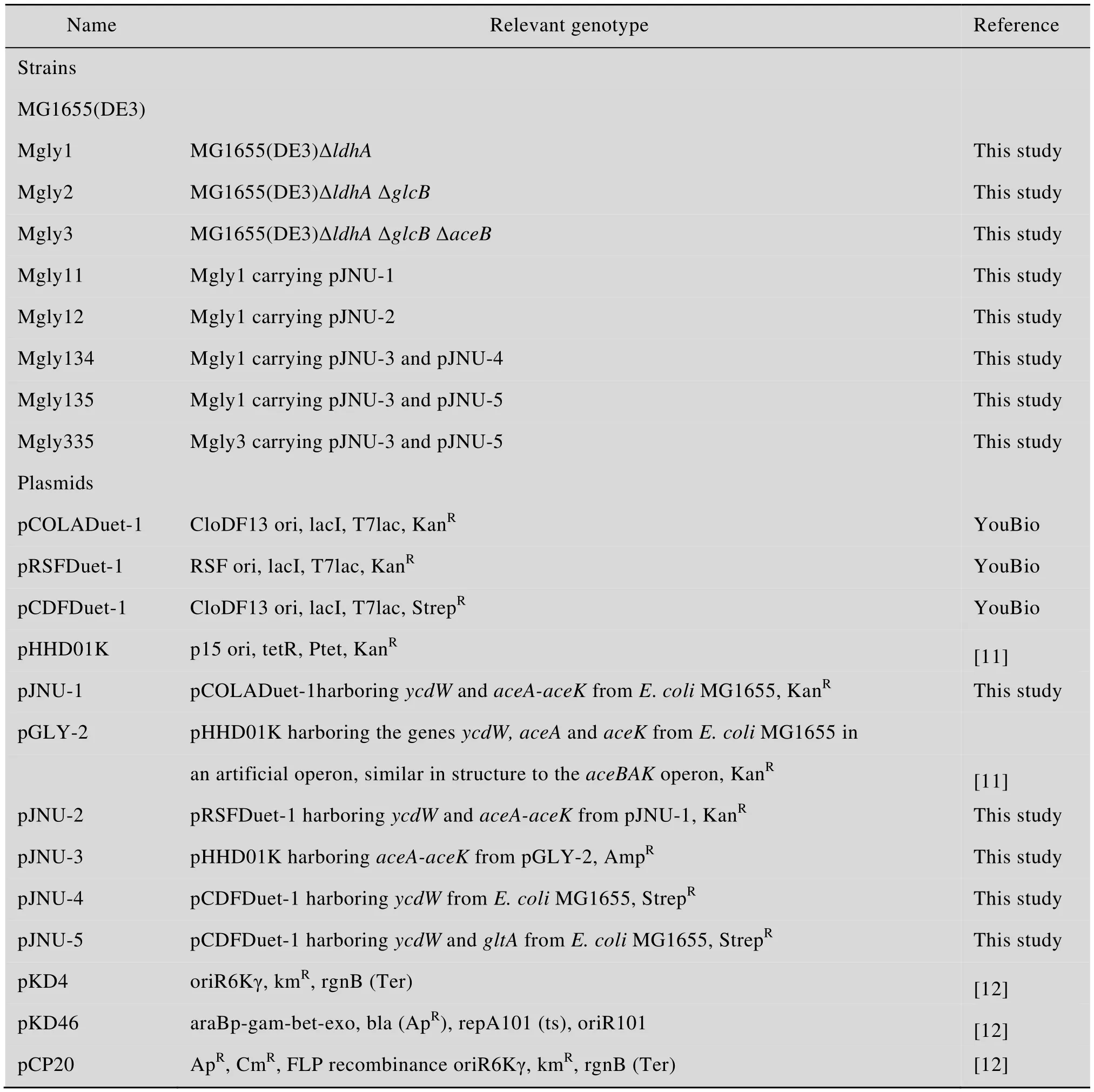
表1 本研究中所用菌株及质粒Table 1 Strains and plasmids used in this study
1.3 重组质粒的构建
重组质粒pJNU-1的构建:利用基因组提取试剂盒提取大肠杆菌 MG1655 (DE3) 基因组,并以此为模板通过pJNU-1-ycdWF和pJNU-1-ycdWR进行 PCR扩增目的基因ycdW[10],37 ℃通过NdeⅠ和XhoⅠ分别双酶切 PCR扩增的基因片段和pCOLDuet-1质粒2 h后,分别用PCR产物纯化试剂盒和胶回收试剂盒回收目的基因和载体片段。T4 DNA连接酶处理目的基因片段与载体片段,16 ℃处理 8-10 h,将上述连接反应液加入100 μL大肠杆菌 JM109转化感受态中冰上放置30 min,42 ℃热激90 s,加入1 mL LB液体培养基,37 ℃摇床孵育 1 h,涂布卡那抗性平板(50 mg/L)。37 ℃培养过夜,通过 veri-pJNU-1 F和veri-pJNU-1 R引物对转化子进行菌落PCR验证。并将阳性菌落转接 LB液体培养基,培养12-16 h,利用质粒小提试剂盒提取质粒,用NdeⅠ和XhoⅠ酶切验证。验证正确的质粒用NcoⅠ和EcoRⅠ在 37 ℃双酶切 2 h,同时用NcoⅠ和EcoRⅠ在37 ℃双酶切PCR扩增的aceAK基因片段2 h。后续连接转化实验步骤同上,用XhoⅠ和EcoRⅠ酶切验证,最后通过生工生物工程 (上海)股份有限公司测序验证。
重组质粒pJNU-2的构建:NcoⅠ和XhoⅠ双酶切质粒pJNU-1,切胶回收片段 (4 356 bp),获得目的基因ycdW和aceAK。NcoⅠ和XhoⅠ酶切质粒pRSFDuet-1,利用胶回收试剂盒回收载体片段。目的基因与载体连接,连接及验证方法同pJNU-1。
重组质粒pJNU-3的构建:以质粒pGLY-2为模板,通过pJNU-3 F和pJNU-3 R引物PCR扩增目的基因aceAK,切胶回收目的基因片段 (3 319 bp),获得目的基因aecAK。EcoRⅠ和BamHⅠ双酶切质粒pGLY-2[10],切胶回收载体片段 (3 592 bp)。利用Gibson组装体系[13]构建重组质粒pJNU-3。验证方法同pJNU-1,验证引物见表2。
重组质粒pJNU-4的构建:NdeⅠ和XhoⅠ双酶切质粒pJNU-1,切胶回收片段(941 bp),获得目的基因ycdW。NdeⅠ和XhoⅠ双酶切质粒pCDFDuet-1,获得载体片段,将目的基因与载体片段连接,构建重组质粒pJNU-4,连接及验证方法同pJNU-1,验证引物见表2。
重组质粒 pJNU-5的构建:以大肠杆菌基因组为模板,PCR扩增目的基因gltA,BamHⅠ和Hind Ⅲ双酶切PCR扩增的基因片段,纯化后获得目的基因片段。BamHⅠ和Hind Ⅲ双酶切质粒pJNU-4,获得载体片段,将目的基因与载体片段连接,连接及验证方法同 pJNU-1,验证引物见表 2。
1.4 宿主菌株MG1655(DE3) 的改造
采用λ-Red重组法[12]敲除了ldhA(乳酸脱氢酶)、glcB、aceB(苹果酸合成酶)。菌落PCR验证基因的正确敲除,所用引物见表2。
具体实施步骤如下。以pKD4为模板,利用引物ko-ldhAF 和ko-ldhAR扩增敲除框,其中包含目的基因两端同源臂各39 bp、2个FRT位点及卡那抗性基因。将转化有pKD46的MG1655(DE3)菌株接于LB培养基中,30 ℃培养过夜,再以1%的接种量转接于50 mL LB培养基,同时加入100 mg/L氨苄青霉素和10 mmol/L阿拉伯糖,30 ℃培养至OD600=0.4-0.6,使pKD46上的3个蛋白充分表达。冰上预冷30 min,5 000 r/min离心收集菌体,然后无菌水离心洗涤1次,10%甘油离心洗涤 2次,将最终菌体浓缩于 500 μL感受态细胞,每管分装100 μL,–80 ℃保藏备用。将纯化后的敲除框10 μL与100 μL感受态细胞充分混匀,转入0.1 cm电转杯中电击转化。电击后迅速加入1 mL LB培养基,37 ℃孵育1 h,涂布卡那抗性平板 (硫酸卡那霉素浓度为35 mg/L),37 ℃培养过夜。然后通过菌落 PCR方法 (验证引物veri-ldhAF和veri-ldhAR) 鉴定阳性克隆。将pCP20电转入阳性克隆,30 ℃培养8 h后,37 ℃过夜培养,诱导FRT重组酶的表达,同时质粒逐渐丢失。在无抗生素的平板上划线,再次进行菌落 PCR验证。相同方法相继敲除glcB和aceB。
1.5 代谢产物的定量检测
1 mL发酵液12 000 r/min离心5 min,取上清用0.22 μm的滤膜过滤,HPLC (Rigol) 定量检测乙醇酸。检测条件如下:色谱柱为AminexHPX-87H(Bio-rad, USA),流动相为 5 mmol/L稀硫酸,流速0.6 mL/min,柱温50 ℃,进样量为20 μL,检测器为示差检测器[10]。

表2 本研究中所用的引物Table 2 List of DNA oligo nucleotide primers used in the cloning of genes and for qPCR
2 结果与分析
2.1 加强乙醛酸循环提高乙醇酸产率
因为乳酸脱氢酶 (ldhA) 催化丙酮酸产生乳酸,从而相对减少了进入TCA的碳流[14],并且乳酸对乙醇酸的定量检测存在干扰,所以在本研究中首先在大肠杆菌MG1655(DE3)中敲除ldhA,获得菌株Mgly1 (MG1655(DE3) ΔldhA),作为本研究的出发菌株。
2.1.1 调节目的基因表达水平,增强乙醛酸循环
由图 1可知,异柠檬酸裂解酶 (aceA) 和乙醛酸还原酶 (ycdW) 为乙醇酸合成途径中的关键酶。异柠檬酸裂解酶 (aceA) 催化异柠檬酸合成乙醛酸,乙醛酸还原酶 (ycdW) 催化乙醛酸合成乙醇酸[15-16]。此外,还有一个与乙醇酸积累密切相关的酶:异柠檬酸脱氢酶激酶/磷酸化酶 (aceK),其可以使异柠檬酸酶磷酸化,从而使异柠檬酸不能转化为α-酮戊二酸,转而生成乙醛酸 (乙醇酸的前体)[17-18]。所以在本研究中首先用T7启动子过量表达以上3个关键基因:ycdW、aceA和aceK,构建质粒pJNU-1 (pCOLADuet-1-ycdW- aceAK),并将其导入Mgly1 (MG1655(DE3) ΔldhA),得到工程菌株Mgly11,其乙醇酸产量为 (1.086±0.151) g/L,产率为 (0.200±0.023) g/g葡萄糖 (理论产率的23.5%)。Mgly11的乙醇酸产量和产率都不高,为了进一步加强乙醛酸循环,改用高拷贝质粒pRSFDuet-1过表达 3个关键基因,构建了质粒pJNU-2 (pRSFDuet-1-ycdW-aceAK),将其转入Mgly1中,获得工程菌株Mgly12,其乙醇酸产量为 (0.953±0.079) g/L,产率为 (0.189±0.017) g/g葡萄糖 (理论产率的 22.2%)。与 Mgly11相比,Mgly12中增加目的基因的拷贝数并没有提高乙醇酸的产率,反而降低了5%。SDS-PAGE分析中发现Mgly11和Mgly12中异柠檬酸裂解酶表达量不足 (图 2),这可能是乙醇酸产率不高的原因之一[19]。Martin等[20]的研究中,利用pTet启动子过量表达乙醇酸合成途径中的 3个基因 (ycdW、aceA、aceK),并积累了1.4 g/L的乙醇酸。所以本研究中利用pTet启动子来过量表达异柠檬酸裂解酶 (aceA) 和异柠檬酸脱氢酶激酶/磷酸化酶(aceK),构建质粒pJNU-3 (pHHD01K-aceAK) 和pJNU-4 (pCDFDuet-1-ycdW) 并将其转入 Mgly1中,获得菌株 Mgly134,其乙醇酸产量为(1.602±0.195) g/L,产率为 (0.24±0.030) g/g葡萄糖 (理论产率的28.2%),较Mgly11提高了20%,但是仍然远低于理论产率。

图1 大肠杆菌中乙醇酸合成途径Fig.1 Glycolate synthesis pathway in Escherichia coli.GLC: glucose; PYR: pyruvate; CIT: citric acid; AKG:α-ketoglutaric acid; SUCC: succinyl-COA; SUC: succinate; FUM: fumaric acid; MAL: malate; OAA: oxaloacetate;GLYX: glyoxylate.
2.1.2 增加进入TCA的碳流,增强乙醛酸
柠檬酸合成酶 (gltA) 作为乙醛酸及三羧酸循环的共用酶,可以催化草酰乙酸和乙酰辅酶 A合成柠檬酸,增加乙酰辅酶A进入三羧酸循环的流量,从而增强三羧酸循环,间接增强支路乙醛酸循环[21-22]。而且在Mgly134分批发酵中有一定乙酸的积累 [(1.051±0.207) g/L],推测在削弱TCA的过程中可能造成乙酰辅酶A的积累,从而导致乙酸的积累。所以在本研究中将gltA过量表达,构建质粒pJNU-5 (pCDFDuet-1-ycdW-gltA),并和 pJNU-3 (pHHD01K-aceAK) 共转入 Mgly1中,得到重组菌株Mgly135,发酵结果如图3所示,其乙醇酸产量为 (2.058±0.144) g/L,产率为(0.326±0.023) g/g葡萄糖 (理论产率的38.3%),Mgly134提高了 35.8%,而且在分批发酵中乙酸积累量明显减少 (图3)。
综上所述,利用pTet启动子过量表达异柠檬酸裂解酶 (aceA) 和异柠檬酸脱氢酶激酶/磷酸化酶 (aceK),同时利用 T7启动子过量表达乙醛酸还原酶 (ycdW),有效增强了乙醇酸合成途径。与单独利用T7启动子过量表达3个目的基因相比,乙醇酸产量提高了 47.5%,产率提高了 20%。在此基础上过量表达柠檬酸合成酶 (gltA),摇瓶发酵中乙醇酸的产量为 2.058 g/L (图 4),产率为0.326 g/g 葡萄糖 (理论产率的38.3%),进一步提高了35.8%。
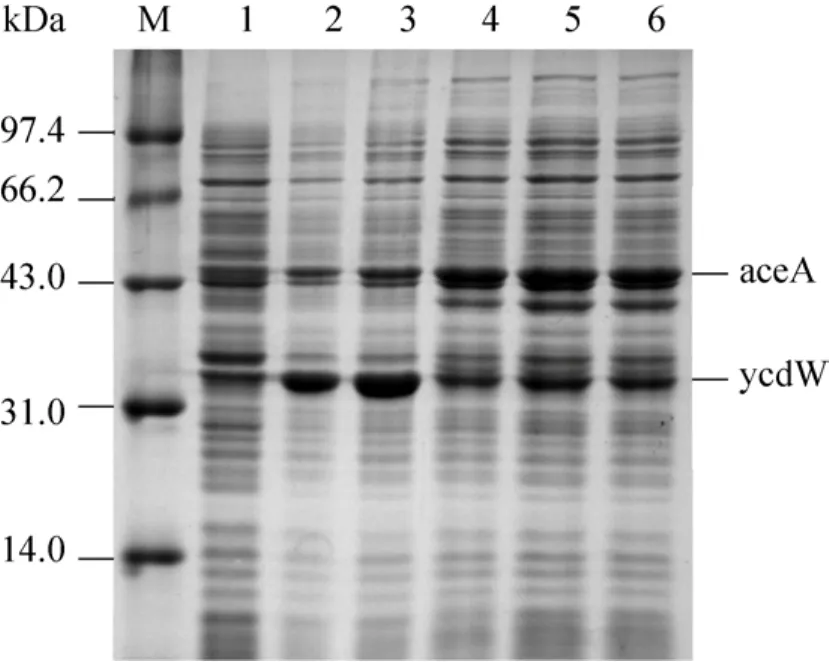
图2 SDS-PAGE比较不同重组菌株中目的蛋白表达水平Fig.2 Comparison of target proteins in different recombinant strains by SDS-PAGE.M: marker; 1: Mgly1;2: Mgly11; 3: Mgly12; 4: Mgly134; 5: Mgly135; 6:Mgly335.Glyoxylate reductase reductase (ycdW):35.4 kDa; Isocitrate lyase (aceA): 47.5 kDa.
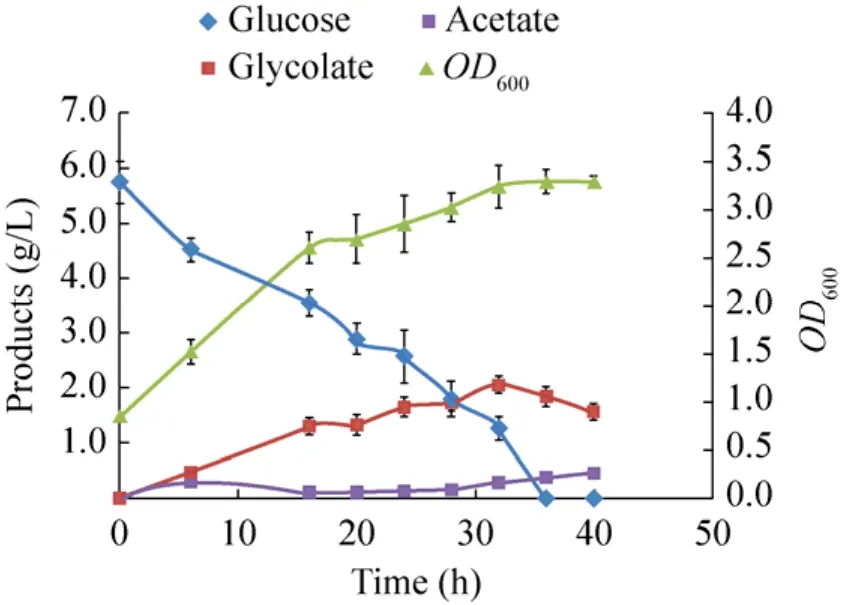
图3 Mgly135摇瓶发酵结果Fig.3 Fermentation results of Mgly335 in shaked flask.Analysis of the cell growth and production of glycolate and acetate of Mgly135 were shown in the figure.And the abscissa means to time after induction.
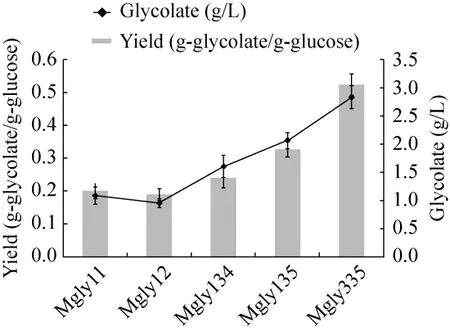
图4 不同重组菌株摇瓶发酵中乙醇酸产量和产率的比较Fig.4 Comparison of glycolic acid production in various metabolite strains.Strains were cultivated in shake flasks with fermentation medium.Final glycolate yield and production of various metabolite strains was displayed using histogram and line chart respectively.
2.2 改造菌株提高乙醇酸产率
在大肠杆菌中,乙醛酸除参与乙醇酸的合成,还可以与乙酰辅酶A在苹果酸合成酶 (glcB和aceB) 催化下生成苹果酸,从而竞争了乙醇酸合成的前体——乙醛酸[23]。Choi等[24]的研究中,以木糖和葡萄糖为碳源,敲除苹果酸合成酶后,乙醇酸产量从1.46 g/L增加到2.31 g/L。因此本研究中,在菌株Mgly1 (MG1655(DE3)ΔldhA) 中敲除了苹果酸合成酶基因glcB和aceB(验证结果如图5所示),获得菌株Mgly3,并将重组质粒pJNU-5 (pCDFDuet-1-ycdW-gltA)和 pJNU-3(pHHD01K-aceAK) 共转入 Mgly3中,得到工程菌株 Mgly335,摇瓶结果中乙醇酸产量为(2.822±0.203) g/L,产率为 (0.522±0.032) g/g葡萄糖(图6),达到理论产率的61.4%,较Mgly135提高了60%。
2.3 工程菌株Mgly335分批发酵生产乙醇酸
2.3.1 Mgly335分批发酵
甘油保藏的菌种于平板上划线活化后,挑取单菌落接种于50 mL的LB液体培养基中,37 ℃、200 r/min摇瓶过夜。以1%接种量将活化后的菌种转接于 60 mL LB液体培养基中,37 ℃、200 r/min培养6 h作为种子备用,以2%的接种量接种于5 L发酵罐中 (装液量为3 L)。发酵条件如下:搅拌转速400 r/min,通气量1 vvm,2 mol/L NaOH维持pH为7.0。
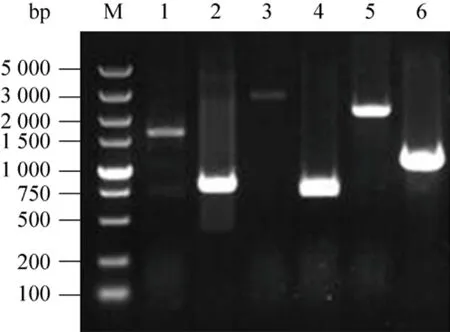
图5 菌落PCR验证基因敲除结果Fig.5 Confirming correct knockout of genes by colony PCR.Confirming correct knockout of ldhA was shown in lane 1 and 2.1: MG1655(DE3) (1 618 bp); 2: MG1655(DE3)ΔldhA (809 bp).Confirming correct knockout of glcB was shown in lane 3 and 4.3: MG1655(DE3) ΔldhA (2 705 bp); 4:MG1655(DE3) ΔldhAΔglcB (2 029 bp).Confirming correct knockout of aceB was shown in lane 5 and 6.5:MG1655(DE3) ΔldhAΔglcB (2 156 bp); 6: MG1655(DE3)ΔldhAΔglcBΔaceB (1 097 bp).
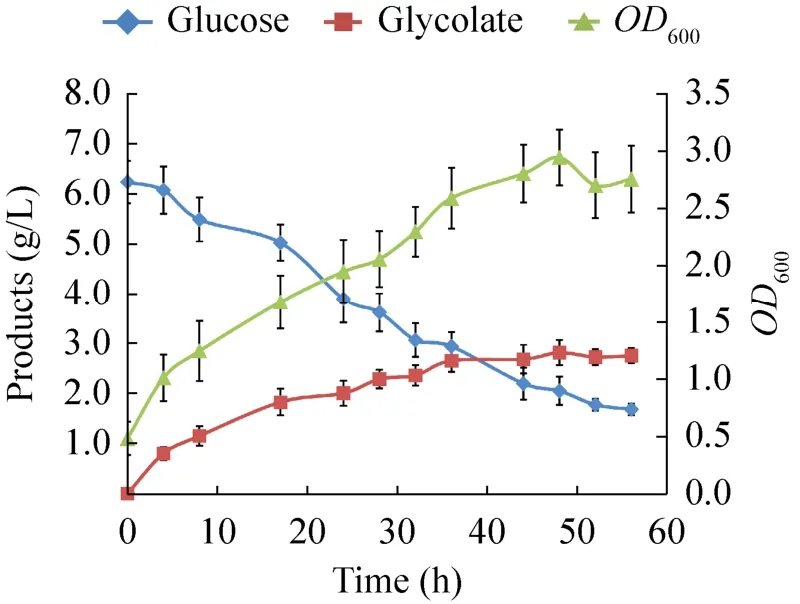
图6 Mgly335摇瓶发酵结果Fig.6 Fermentation results of Mgly335 in shaked flask.Analysis of the cell growth and production of glycolate of Mgly335 were shown in the figure.The yield of glycolic acid of Mgly335 reached 0.522 g/g glucose,which was up to 61.4% of the theoretical maximum yield from glucose of 0.85 g/g glucose.And the abscissa means to time after induction.
接种后37 ℃培养至OD600为0.8左右时,将发酵温度降到30 ℃,加入250 ng/mL aTc (脱水四环素) 和1 mmol/L IPTG诱导目的基因表达,发酵结果如图 7A所示。加入诱导剂后乙醇酸开始积累,接种29 h后,葡萄糖耗尽,乙醇酸产量达到最大为3.888 g/L,产率为0.474 g/g 葡萄糖,并且在发酵过程中无乙酸等杂酸的积累。
2.3.2 Mgly335补料分批发酵
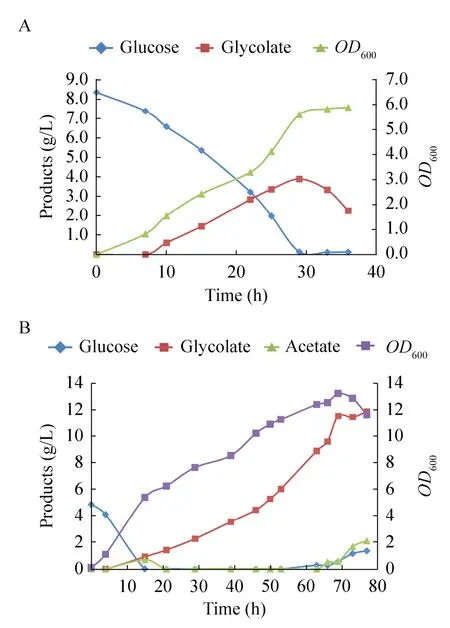
图7 Mgly335分批发酵结果Fig.7 The results of Mgly335 in batch fermentation.Analysis of the cell growth and production of Mgly335 in a 5 L bioreactor.The production of glycolic acid reached 3.888 g/L with 0.474 g/g glucose yield in batch fermentation and 11.909 g/L with 0.496 g/g glucose in fed-batch, which were shown in A and B respectively.
种子液制备和发酵条件同分批发酵,发酵结果如图7B所示,接种6 h后加入诱导剂,此阶段细胞处于对数期,比生长速率较高,乙醇酸和乙酸基本同步积累。接种后 15 h时葡萄糖耗尽,开始进行恒速补料,补料速度为0.3 g/(L·h),细胞开始处于“饥饿”状态,在补入的葡萄糖耗尽的同时细胞开始以已经积累的乙酸作为碳源,乙酸在21 h时被耗尽,同时细胞的比生长速率较之前略有降低。在63 h时菌体量达到峰值,之后菌体量开始缓慢下降,表示细胞生长进入衰亡期。在这一阶段,由于细胞不再继续生长和繁殖,代谢量也相应减少,葡萄糖和乙酸开始积累,而乙醇酸产量的增速也明显减慢,在 77 h时达到最大值11.909 g/L,产率为 0.496 g/g 葡萄糖 (理论产率的 58.3%)。
3 讨论
乙醇酸是分子量最小的果酸,它在化学清洗、生物降解、日用化工等行业得到了广泛的应用,其需求量也在逐年增加。全生物方法合成乙醇酸与化学方法相比,具有反应条件温和、对环境影响小等优点,因此成为现在研究的热点。Cam等[6]在大肠杆菌中利用木糖为底物获得4.3 g/L乙醇酸,产率为0.43 g/g木糖。Alkim[7]通过改造大肠杆菌的木酮糖磷酸途径和乙醛酸循环,以木糖和葡萄糖为混合碳源获得3.73 g/L乙醇酸,产率为0.497 g乙醇酸/g糖。而以葡萄糖为底物较木糖相比合成乙醇酸具有底物廉价、产品纯度高等突出优点。乙醇酸的生物合成途径包括3个关键酶 (异柠檬酸裂解酶、乙醛酸还原酶和异柠檬酸脱氢酶激酶/磷酸化酶) 和两个关键反应 (异柠檬酸裂解生成乙醛酸和乙醛酸转化为乙醇酸)。Dhamankar 等[11]在大肠杆菌 MG1655(DE3) 中以葡萄糖为底物,利用 T7启动子过量表达乙醇酸合成途径中的 3个关键基因 (ycdW、aceA、aceK) 实现了乙醇酸的积累,产量为0.7 g/L,产率为0.0875 g/g葡萄糖;而利用pTet启动子过量表达3个基因,获得乙醇酸产量为1.43 g/L,产率为0.178 g/g葡萄糖,低于理论产率 (0.85 g/g葡萄糖)。本研究中利用不同拷贝数的表达质粒和更换启动子的方法共同调节上述 3个关键基因的表达水平,用强启动子 pTet过量表达aceAK,提高异柠檬酸裂解酶的表达量,同时用T7启动子过量表达ycdW(乙醛酸还原酶),乙醇酸产量为1.602 g/L,产率为0.24 g/g葡萄糖,虽然乙醇酸产量值略高于上述文献报道,但是其产率却明显提高,这说明在生物法合成乙醇酸的过程中过量表达关键基因的同时,调节3个关键酶的表达水平和平衡两步关键反应的速率也是提高乙醇酸产率的重要因素。因此,本研究为提高乙醇酸产量和产率提供了新策略,并可将此策略应用于提高其他有机酸的生物合成。
本研究在上述基础过量表达柠檬酸合成酶编码基因 (gltA),结果显示工程菌株Mgly135获得了2.058 g/L乙醇酸,产率为0.326 g/g 葡萄糖 (理论产率的38.3%),乙醇酸产率提高了36%,这表明直接调节乙醛酸循环并不是唯一提高乙醇酸产率的策略,可以通过加强三羧酸循环进而增加乙醛酸循环的代谢流,从而实现乙醇酸产率的提高。这也为进一步的研究提供新的思路,即可以通过改善前体通路进而提高乙醇酸的产率和合成效率。
Choi等[24]的研究中,敲除苹果酸合成酶编码基因 (glcB和aceB) 后,以木糖和葡萄糖为混合碳源进行发酵培养后,乙醇酸产量从1.46 g/L增加到 2.31 g/L。本研究在敲除乳酸脱氢酶(ldhA)、过量表达乙醇酸合成途径中的 3个关键基因 (ycdW、aceA、aceK) 和过量表达柠檬酸合成酶编码基因 (gltA) 的基础上进一步敲除了苹果酸合成酶编码基因 (glcB和aceB),得到工程菌株 Mgly335,以葡萄糖为唯一碳源进行发酵培养后,乙醇酸产量为2.822 g/L,产率为0.522 g/g葡萄糖,进一步提高了乙醇酸的产量和产率。这说明阻断旁路代谢途径的同时减少乙醇酸合成前体——乙醛酸的消耗,能够增加进入乙醇酸合成途径的碳流,进一步提高了乙醇酸的产量及产率。同时这一结果也提示可能还有其他旁路代谢途径对乙醇酸的合成产生影响,因此,后续实验中我们将对乙醇酸及其前体乙醛酸的其他代谢途径作进一步研究[25]。
Mgly335工程菌补料分批发酵后,发酵77 h时乙醇酸产量达到最大值 11.909 g/L,产率为0.496 g/g葡萄糖 (理论产率的58.3%),使乙醇酸产量大幅度提高,为乙醇酸的规模化生产提供了基础。分批发酵中乙醇酸的产率较摇瓶发酵略低,所以在后续研究中,我们将探究各种发酵影响因素[26],如不同碳源、氮源、溶氧、pH、诱导时间、微量元素含量等对乙醇酸产量和产率的影响,以得到更高的乙醇酸产量和产率,并将改进后的方法用于工业化生产,以便节约生产成本,提高生产效率,减少生产对环境的影响。
[1]Koivistoinen OM, Kuivanen J, Barth D, et al.Glycolic acid production in the engineered yeastsSaccharomyces cerevisiaeandKluyveromyceslactis.Microb Cell Fact,2013, 12: 82.
[2]Kataoka M, Sasaki M, Hidalgo AR, et al.Glycolic acid production using ethylene glycol-oxidizing microorganisms.Biosci Biotechnol Biochem, 2001, 65(10): 2265–2270.
[3]Shi YH, Sun HY, Lu DM, et al.Separation of glycolic acid from glycolonitrile hydrolysate by reactive extraction with tri–n–octylamine.Sep Purif Technol,2006, 49(1): 20–26.
[4]He YC, Xu JH, Su JH, et al.Bioproduction of glycolic acid from glycolonitrile with a new bacterial isolate ofAlcaligenessp.ECU0401.Appl Biochem Biotechnol,2010, 160(5): 1428–1440.
[5]Ñancucheo I, Johnson DB.Production of glycolic acid by chemolithotrophic iron- and sulfur-oxidizing bacteria and its role in delineating and sustaining acidophilic sulfide mineral-oxidizing consortia.Appl Environ Microb, 2010, 76(2): 461–467.
[6]Cam Y, Alkim C, Trichez D, et al.Engineering of a synthetic metabolic pathway for the assimilation of(D)-xylose into value-added chemicals.ACS Synth Biol,2016, 5(7): 607–618.
[7]Alkim C, Trichez D, Cam Y, et al.The synthetic xylulose-1 phosphate pathway increases production of glycolic acid from xylose-rich sugar mixtures.Biotechnol Biofuels, 2016, 9: 201.
[8]Zahoor A, Otten A, Wendisch VF.Metabolic engineering ofCorynebacteriumglutamicumfor glycolate production.J Biotechnol, 2014, 192: 366–375.
[9]Pereira B, Li ZJ, de Mey M, et al.Efficient utilization of pentoses for bioproduction of the renewable two-carbon compounds ethylene glycol and glycolate.Metab Eng,2016, 34: 80–87.
[10]Dischert W, Figge R, Soucaille P.Use of inducible promoters in the production of glycolic acid: US,20130089903.2013-04-11.
[11]Dhamankar H, Tarasova Y, Martin CH, et al.EngineeringE.colifor the biosynthesis of 3-hydroxy-γ-butyrolactone(3HBL) and 3, 4-dihydroxybutyric acid (3, 4-DHBA) as value-added chemicals from glucose as a sole carbon source.Metab Eng, 2014, 25: 72–81.
[12]Datsenko KA, Wanner BL.One-step inactivation of chromosomal genes inEscherichiacoliK-12 using PCR products.Proc Natl Acad Sci USA, 2000, 97(12):6640–6645.
[13]Gibson DG, Young L, Chuang RY, et al.Enzymatic assembly of DNA molecules up to several hundred kilobases.Nat Methods, 2009, 6(5): 343–345.
[14]Bunch PK, Mat-Jan F, Lee N, et al.TheldhAgene encoding the fermentative lactate dehydrogenase ofEscherichia coli.Microbiol, 1997, 143(1): 187–195.
[15]Ogawa T, Murakami K, Mori H, et al.Role of phosphoenolpyruvate in the NADP-isocitrate dehydrogenase and isocitrate lyase reaction inEscherichiacoli.J Bacteriol, 2007, 189(3): 1176–1178.
[16]Maloy SR, Nunn WD.Genetic regulation of the glyoxylate shunt inEscherichia coliK-12.J Bacteriol,1982, 191(13): 4251–4258.
[17]Nuñez MF, Pellicer MT, Badia J, et al.Biochemical characterization of the2-ketoacid reductases encoded by ycdW and yiaE genes inEscherichia coli.Biochem J,2001, 354(3): 707–715.
[18]Zheng JM, Jia ZC.Structure of the bifunctional isocitrate dehydrogenase kinase/phosphatase.Nature,2010, 465(7300): 961–965.
[19]Matsuoka M, McFadden BA.Isolation, hyperexpression,and sequencing of the aceA gene encoding isocitrate lyase inEscherichiacoli.J Bacteriol, 1988, 170(10):4528–4536.
[20]Martin CH, Dhamankar H, Tseng HC, et al.A platform pathway for production of 3-hydroxyacids provides a biosynthetic route to 3-hydroxy-γ- butyrolactone.Nat Commun, 2013, 4: 1414.
[21]Li YJ, Huang B, Wu H, et al.Production of succinate from acetate by metabolically engineeredEscherichia coli.ACS Synth Biol, 2016, 5(11): 1299–1307.
[22]Walsh K, Koshland DE Jr.Characterization of rate-controlling stepsin vivoby use of an adjustable expression vector.Proc Natl Acad Sci USA, 1985,82(11): 3577–3581.
[23]Vanderwinkel E, de Vlieghere M.Physiologie et génétique de l'isocitritase et des malate synthases chezEscherichiacoli.Eur J Biochem, 1968, 5(1): 81–90.
[24]Choi SY, Park SJ, Kim WJ, et al.One-step fermentative production of poly (lactate-co-glycolate) from carbohydrates inEscherichiacoli.Nat Biotechnol, 2016,34(4): 435–440.
[25]Núñez MF, Pellicer MT, Badía J, et al.The geneyghKlinked to the glc operon ofEscherichia coliencodes a permease for glycolate that is structurally and functionally similar to L-lactate permease.Microbiol,2001, 147(4): 1069–1077.
[26]Sohoni SV, Nelapati D, Sathe S, et al.Optimization of high cell density fermentation process for recombinant nitrilase production inE.coli.Bioresour Technol, 2015,188: 202–208.
猜你喜欢
成都医学院学报(2022年4期)2022-08-19
江西水产科技(2022年2期)2022-05-17
城市道桥与防洪(2022年3期)2022-05-08
现代仪器与医疗(2021年1期)2021-06-09
江西农业学报(2021年4期)2021-04-20
安全与环境工程(2021年2期)2021-04-02
煤炭加工与综合利用(2020年6期)2020-07-17
三农资讯半月报(2020年11期)2020-06-21
益寿宝典(2018年8期)2018-10-22
妇女之友(2017年8期)2017-10-24
